
Selective expression of neurodegenerative diseases-related mutant p150Glued in midbrain dopaminergic neurons causes progressive degeneration of nigrostriatal pathway


Abstract
Aim: Missense mutations of dynactin subunit p150Glued have been associated with multiple neurodegenerative diseases, including Perry syndrome, characterized by inherited parkinsonism, depression, weight loss, and hypoventilation. The current study investigated how the pathogenic mutant p150Glued affects the integrity and function of the nigrostriatal dopaminergic (DA) pathway in vivo.
Methods: Using a tetracycline-controlled transcriptional regulation system, transgenic mouse models were generated with selective overexpression of wild-type, motor neuron disease-related G59S mutant, or Perry syndrome-related G71R mutant human p150Glued in midbrain DA neurons. A series of behavioral, neuropathological, neurochemical, electrochemical, and biochemical studies were performed on the mice to examine and compare the pathogenic impact of the two mutant p150Glued on the survival and function of midbrain DA neurons.
Results: Compared with non-transgenic control mice, transgenic mice overexpressing wild-type human p150Glued showed neither motor phenotypes nor pathological, functional, or biochemical abnormalities of the nigrostriatal DA pathway. Transgenic mice overexpressing G59S mutant p150Glued displayed weight loss, motor deficits, early-onset defects in dopamine transmission, and early-onset loss of DA neurons and axons. Transgenic mice overexpressing G71R p150Glued mutant exhibited hyperactivities, impaired motor coordination, early-onset dysfunction of dopamine uptake, and late-onset loss of DA neurons and axons. In addition, overexpression of either G59S or G71R mutant p150Glued in midbrain DA neurons preferentially downregulated the expression of dopamine transporter at dopaminergic axon terminals. Furthermore, G59S mutant p150Glued rather than G71R mutant p150Glued formed aggregates in midbrain DA neurons in vivo, and the aggregates trapped dynein/dynactin, co-localized with lysosomes, and upregulated ubiquitination.
Conclusion: These findings demonstrate that selective expression of either G59S or G71R mutant p150Glued in mouse midbrain DA neurons leads to progressive degeneration of the nigrostriatal DA pathway and indicate that G59S and G71R mutant p150Glued exhibit differential pathogenic impact on the survival and function of midbrain DA neurons in vivo.
Keywords
INTRODUCTION
Cytoplasmic dynein and its cofactor dynactin mediate microtubule minus-end-directed trafficking, performing critical roles in mitosis, intracellular positioning and movement of organelles, retrograde axonal transport, and synapse formation and stability[1-3]. Dynactin is a macromolecular protein complex consisting of more than ten types of subunits, with the largest subunit p150Glued binding directly with dynein and microtubule[4]. Missense mutations of DCTN1, the gene encoding p150Glued, have been linked to different neurodegenerative diseases, in which the G59S missense mutation was linked to autosomal dominant motor neuron disease, and 13 more mutations (F52L, K56R, G67D, K68E, G71A/R/E/V, T72P, Q74P, Y78C/H, and Q93H) were associated with Perry syndrome[5-8]. Perry syndrome is an autosomal dominant neurodegenerative disorder characterized by a series of symptoms (parkinsonism, depression, central hypoventilation, and weight loss) and pathology of severe neuronal loss in the substantia nigra and locus coeruleus[9,10]. However, the underlying pathogenic mechanism and potential therapeutic target of neurodegeneration in Perry syndrome remain elusive.
P150Glued contains two microtubule-binding domains (MTBDs) at its N-terminus: the cytoskeleton-associated protein and glycine-rich (CAP-Gly) domain, which interacts with both tubulin and microtubule end binding proteins, and the adjacent serine-rich basic domain, which has an affinity for tubulin[11,12]. The MTBDs of p150Glued are required to initiate retrograde transport and maintain microtubule stability in axons[13-15]. Deleting the MTBDs in p150Glued upregulates microtubule acetylation, increases the surface level of ionotropic glutamate receptors, disrupts neuromuscular junctions, and leads to selective degeneration of spinal motor neurons[16]. Motor neuron disease-related G59S mutation of p150Glued and all the Perry syndrome-related mutations are point mutations clustered within or close to the CAP-Gly domain, resulting in loss-of-function of the MTBD[6-8]. Furthermore, G59S mutant p150Glued, as well as G71A, G71E, G71R, T72P, Q74P, and F52L mutant p150Glued, form cytoplasmic aggregates in vitro in a time- and concentration-dependent manner[17-19], suggesting both loss-of-function and toxic-gain-of-function mechanisms might be involved in DCTN1 mutation-induced neurodegeneration. Additionally, G59S mutant p150Glued is less stable[20], and both G59S and Perry syndrome-related mutant p150Glued compromise normal functions of dynactin p150Glued in a dominant-negative fashion when overexpressed[13,14,21], suggesting a dominant-negative mechanism for DCTN1 mutation-induced neurodegeneration. Movement disorder and the underlying neuropathology, including loss of spinal motor neurons and degeneration of axon and neuromuscular junctions, have been reported in G59S mutant p150Glued knock-in and transgenic mice[20-22]. Behavioral defects have also been found in G71A mutant p150Glued knock-in and transgenic mice[23,24]. However, how the pathogenic mutant p150Glued affects the integrity and function of the nigrostriatal DA pathway has not been critically examined in animal models.
Here, we investigated and compared the pathogenic impact of two different neurodegenerative diseases-related mutant p150Glued in the survival and function of midbrain DA neurons in vivo. Transgenic mouse models with selective overexpression of wild-type, G59S mutant, or G71R mutant human p150Glued in midbrain DA neurons were generated using a tetracycline-controlled transcriptional regulation system. A series of experiments were performed to identify any behavioral, neuropathological, neurochemical, electrochemical, and biochemical abnormalities in the transgenic mice. Our findings highlight the importance of p150Glued and its MTBDs in maintaining the survival and function of midbrain DA neurons and reveal the pathogenic similarity and difference between G59S and G71R mutant 150Glued.
METHODS
Animals
The tetO-human DCTN1 WT transgenic mice (for simplicity, referred to as tetO-WT) carrying the wild-type (WT) human p150Glued gene (DCTN1) under the control of tetracycline operator (tetO) were generated as described previously[25,26]. The tetO-WT mice were mated with Pitx3-IRE2-tTA mice (referred to as tTA, a “Tet-Off” tool, JAX stock #021962), which predominantly express tetracycline-controlled transactivator protein (tTA) in midbrain DA neurons[27]. This breeding strategy produced Pitx3-IRE2-tTA/tetO-human DCTN1 WT transgenic mice (referred to as WT Tg), which had the selective expression of wild-type human p150Glued in midbrain DA neurons, and littermate controls [non-transgenic (nTg), tetO-WT, and tTA mice].
Following the same protocol, Pitx3-IRE2-tTA/tetO-human DCTN1 G59S transgenic mice (referred to as G59S Tg), which had the selective expression of G59S mutant human p150Glued in midbrain DA neurons, and littermate controls (nTg, tetO-G59S, and tTA mice) were obtained.
Following the same protocol, Pitx3-IRE2-tTA/tetO-human DCTN1 G71R transgenic mice (referred to as G71R Tg), which had the selective expression of G71R mutant human p150Glued in midbrain DA neurons, and littermate controls (nTg, tetO-G71R, and tTA mice) were obtained.
The mice were housed in a 12 h light/dark cycle and fed a regular diet ad libitum. All mouse work followed the guidelines approved by the Institutional Animal Care and Use Committees of National Institute on Aging, NIH.
Genotyping
Genomic DNA was extracted from tail biopsy using DirectPCR Lysis Reagent (Viagen Biotech) and used for PCR amplification. Genotyping was performed with specific sets of PCR primers for Pitx3-IRES2-tTA transgene (GACTGGCTTGCCCTCGTCCCA and GTGCACCGAGGCCCCAGATCA)[27] and tetO-human DCTN1 transgene (AGGAGATGGCTGATACTGCTGATG and TTCATCGCTTCCAAGTCTCCCACA).
Behavior tests
Cohorts of WT Tg mice and littermate controls [nTg (n = 10), tTA (n = 10), tetO-WT (n = 10), and WT Tg (n = 10)] were repeatedly weighed and assessed for motor function at 1, 6, 12, and 18 months of age.
Cohorts of G59S Tg mice and littermate controls [nTg (n = 10), tTA (n = 11), tetO-G59S (n = 10), and G59S Tg (n = 10)] were repeatedly weighed and assessed for motor function at 1, 6, 12, and 18 months of age.
Cohorts of G71R Tg mice and littermate controls [nTg (n = 11), tTA (n = 10), tetO-G71R (n = 10), and G71R Tg (n = 10)] were repeatedly weighed and assessed for motor function at 1, 6, 12, and 18 months of age.
Open-field test. Mice were placed in the open-field apparatus of the Flex-Field Activity System (San Diego Instruments). Ambulatory, rearing, and fine movements of mice were traced and quantified with infrared photobeams sensors and Flex-Field software in the unit as the number of beam breaks per 30 min.
Rotarod test. Mice were placed onto a rotating rod with auto-acceleration from 0 to 40 rpm within 1 min (San Diego Instruments). The time length that each mouse stayed on the rotating rod was measured across three trials.
Histology, immunostaining, and light microscopy
Mice were sacrificed and perfused with 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS). Mouse brains were dissected, post-fixed in 4% PFA/PBS solution overnight, and submerged in 30% sucrose in PBS for at least 72 h. The 40 μm-thick brain sections were prepared using CM1950 cryostat (Leica) and stained with antibodies specific to human and mouse p150Glued (amino acid 3-202 at the N-terminus of p150Glued, 1:200, BD Biosciences, Catalog # 610474, recognizing wild-type p150Glued but not G59S mutant p150Glued, G71R mutant p150Glued, or p135+), human and mouse p150Glued and p135+ (amino acid 1266-1278 at the C-terminus of p150Glued and p135+, 1:500, Abcam, Catalog # ab11806, recognizing wild-type, G59S mutant and G71R mutant p150Glued, and p135+), tyrosine hydroxylase (TH, 1:2500, Pel-freez, Catalog # P40101-150; 1:500, ImmunoStar, Catalog # 22941), dynactin subunit p50 (1:1000, Millipore, Catalog # AB5869P), dynactin subunit actin-related protein 1 (ARP1, 1:1000, Millipore, Catalog # AB6058), cytoplasmic dynein light intermediate chain (DLIC, 1:500, Abcam, Catalog # ab123901), lysosomal associated membrane protein 2 (LAMP2, 1:500, Abcam, Catalog # ab13524), and ubiquitin (1:500, Enzo Life Sciences, Catalog # BML-PW0930), as suggested by manufacturers. Fluorescent signals were visualized with Alex Fluor-conjugated secondary antibody (1:500, Invitrogen, Catalog # A-11055, A-21202, A-21206, A-21208, A-21447, A10036, and A10040) and examined using LSM 880 laser-scanning confocal microscope (Zeiss). The paired images in all figures were collected using identical gain and offset settings, processed uniformly, and displayed as either a single optic layer or the maximum-intensity projections of the z-series confocal stack at 1.0 μm intervals.
Stereology
The number of midbrain dopaminergic neurons was estimated by unbiased stereology, as described previously[27,28]. According to Paxinos and Franklin’s The Mouse Brain in Stereotaxic Coordinates (4th edition), a series of coronal sections (40 μm thick) across the midbrain (every fourth section from Bregma -2.54 to -4.24 mm, 10 sections per animal) were stained with an antibody specific to TH (1:2500, Pel-Freez, Catalog # P40101-150). After subsequent staining with Vectastain Elite ABC Kit, sections were visualized with DAB Kit (Vector Laboratories). Bright-field images were captured, and the number of TH-positive neurons was assessed using the optical fractionator function of Stereo Investigator 10 (MicroBrightField) under the Axio microscope Imager A1 (Zeiss). Counters were blinded to the genotypes of mice, and the sampling scheme was designed to have a coefficient of error less than 10% for the reliability of results.
Image analysis
Quantitative analysis of images was performed with ImageJ (NIH). After converting image type to 8-bit, areas of interest were selected with Freehand tools, and then mean optical intensities or area fractions were measured. The mean intensity for the background area was subtracted from the selected area to determine the net mean intensity. For quantitative analysis of DA neurite dystrophy, 5 tile images per animal (5 sections per animal and 1 tile image per section) from the substantia nigra pars reticulata (SNr) were taken with a 20× lens. Dystrophic DA neurites were defined as neuritic varicosity ≥ 25 μm2. The area of dystrophic TH-positive DA neurites and the area of the SNr were quantified with ImageJ (NIH). For quantitative analysis of DA axon degeneration, 50 images per animal (10 sections per animal and 5 images per section) from the dorsal striatum were taken with a 63× lens. The area fraction of TH-positive DA axon terminals in each image was quantified with ImageJ (NIH). For quantitative analysis of G59S mutation-induced p150Glued aggregates, aggregates were defined as puncta ≥ 1 μm2.
High-performance liquid chromatography (HPLC)
The dorsal striatum of mice was dissected, weighed, and homogenized by sonication in 0.1 N perchloric acid containing 100 μM EDTA (500 μl buffer per 100 mg tissue). After centrifugation, the supernatant was collected and assayed for dopamine and DOPAC content by HPLC with electrochemical detection, as described previously[27,28]. Briefly, the mobile-phase solution containing octanesulfonic acid was used as an ion-pairing agent and pumped isocratically through a reversed-phase liquid chromatographic column. DA and DOPAC were measured by the current produced after exposure of the eluate to a flow-through electrode set to oxidizing and then reducing potentials in series, with recordings from the last electrode reflecting reversibly oxidized species.
Fast-scan cyclic voltammetry (FSCV)
The kinetics of striatal dopamine release evoked by electrical stimulation was measured using FSCV, as described previously[27,28]. The 400 μm-thick slices of dorsal striatum were prepared and transferred into
Western blot analysis
Mouse brain tissues were homogenized and sonicated in 1% SDS lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 2 mM EDTA, pH 7.5, and 1% SDS) supplemented with Protease/Phosphatase Inhibitor Cocktails (ThermoFisher Scientific). Lysates were centrifuged at 15,000 g for 15 min at 4 °C. Supernatants were collected, and protein content was quantified with the bicinchoninic acid (BCA) assay kit (ThermoFisher Scientific). Equal amounts of total proteins were size-fractioned with NuPage 4%-12% Bis-Tris gel in MES or MOPS running buffer (ThermoFisher Scientific). After transferring to nitrocellulose membranes with the Trans-Blot Turbo Transfer System (Bio-Rad), proteins were immunoblotted with specific primary antibodies to human p150Glued (1:1000, Covance, custom-made), human and mouse p150Glued and p135+ (1:1000, Abcam, Catalog # ab11806), dynactin subunit DCTN4 (1:1000, Abcam, Catalog # ab170107), dynactin subunit p50 (1:1000, BD Biosciences, Catalog # 611002), dynactin subunit actin-related protein 1 (ARP1, 1:1000, Sigma-Aldrich, Catalog # A5601), tyrosine hydroxylase (TH, 1:1000, Sigma-Aldrich, Catalog # T1299), dopamine transporter (DAT, 1:1000, Millipore, Catalog # MAB369), and β-actin (1:5000, Sigma-Aldrich, Catalog # A1978). Protein signals were visualized using the Odyssey System with IRDye secondary antibodies (LI-COR Biosciences, Catalog # 926-32212, 926-32219, 926-3221, 926-32411, and 926-68023) and quantified with NIH ImageJ software.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 9 (GraphPad Software). Data are presented as mean ± SEM. Statistical significance was determined by comparing means of different groups using unpaired t-test, one-way, and two-way ANOVA with post hoc analysis. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
RESULTS
Generation of mouse models with selective expression of wild-type or mutant human p150Glued in midbrain DA neurons
To investigate the involvement and mechanism of p150Glued in neurodegeneration in vivo, three lines of tetO-human DCTN1 mice (tetO-WT, tetO-G59S, or tetO-G71R) with inducible expression of wild-type or mutant human p150Glued under the transcriptional control of tetracycline operator (tetO) were generated. The tetO-human DCTN1 mice were then crossed with Pitx3-IRE2-tTA knock-in mice to obtain Pitx3-IRE2-tTA/tetO-human DCTN1 WT (WT Tg), Pitx3-IRE2-tTA/tetO-human DCTN1 G59S (G59S Tg), or Pitx3-IRE2-tTA/tetO-human DCTN1 G71R (G71R Tg) bigenic mice [Figure 1A]. Since the tetracycline transactivator (tTA) is specifically expressed in midbrain DA neurons under the endogenous Pitx3 promoter[27], it turned on the expression of human wild-type or mutant p150Glued selectively in midbrain DA neurons [Figure 1A]. The target overexpression of human p150Glued in midbrain DA neurons of one-month-old WT mice was visualized by immunostaining [Figure 1B]. Using a human p150Glued-specific antibody, Western blot confirmed the expression of human p150Glued in the midbrain homogenates of one-month-old WT Tg, G59S Tg, and G71R Tg mice but not nTg mice [Figure 1C and D]. The steady level of G59S human p150Glued was significantly less (approximately 30%) compared with WT or G71R human p150Glued. Western blot further revealed substantially increased levels of total (human and mouse) p150Glued (approximately two-fold) and other dynactin subunits, including DCTN4, p50, and ARP1, but markedly reduced level of p135+ (alternative splicing isoforms of Dctn1 lacking the N-terminus) in the midbrain homogenates of WT Tg, G59S Tg, and G71R Tg mice compared to the nTg mice [Figure 1C and D].
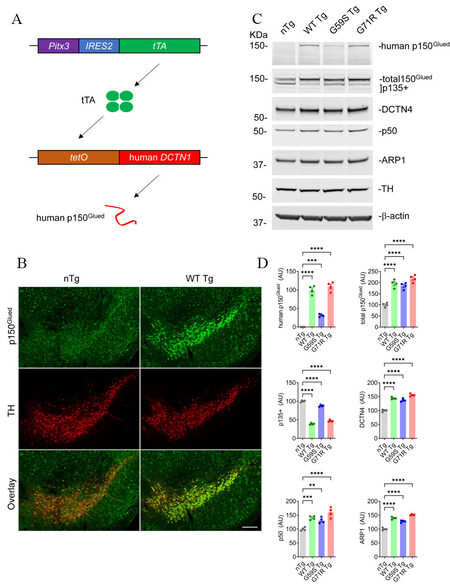
Figure 1. Selective expression of wild-type or mutant human p150Glued in mouse midbrain DA neurons. (A) The schematic diagram depicts the generation of transgenic mice with target expression of human p150Glued in midbrain DA neurons by crossing Pitx3-IRE2-tTA and tetO-human DCTN1 mice. (B) Immunofluorescent images show the staining of p150Glued (green) and TH (red) in the coronal midbrain sections of one-month-old nTg and WT Tg mice. DA neurons were visualized by TH staining. Scale bar: 200 μm. (C, D) Western blots show the expression of human p150Glued, total (human and mouse) p150Glued, p135+, DCTN4, p50, and ARP1 in the midbrain homogenates of one-month-old nTg, WT Tg, G59S Tg, and G71R Tg mice. β-actin and TH were used as the loading control. The bar graph estimates the protein level of dynactin subunits normalized with β-actin (n = 4 per genotype). Data are presented as mean
G59S Tg and G71R Tg mice exhibit profound motor abnormalities.
As motor symptoms and weight loss are the main clinical manifestations in familial motor neuron disease and Perry syndrome cases with DCTN1 mutation[5], WT Tg, G59S Tg, G71R Tg mice, and their littermate controls were weighed, tested for locomotion activity in the open-field test, and examined for motor coordination in the rotarod test at 1, 6, 12, and 18 months of age. WT Tg mice developed normally and exhibited comparable body weight and motor function with littermate single- or non-transgenic mice in the open field test and the rotarod test [Figure 2A-D]. G59S Tg mice displayed substantial weight loss starting at 12 months of age, normal ambulatory movement, dramatic reduction of rearing movement at 18 months of age, and significant impairment in rotarod test starting at six months of age [Figure 2E-H]. G71R Tg mice showed normal body weight, significant hyperactivities of ambulatory movement and rearing, and substantially impaired rotarod performance starting at six months of age [Figure 2I-L]. Together, these findings demonstrate that selective expression of either G59S or G71R mutant p150Glued in midbrain DA neurons causes profound motor abnormalities in mice and suggest that both G59S and G71R mutant p150Glued may impair the function and survival of midbrain DA neurons.
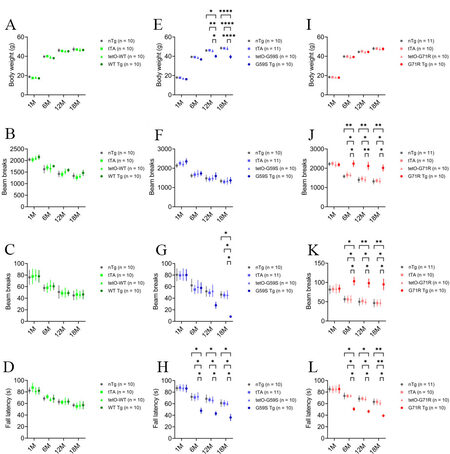
Figure 2. Profound motor abnormalities of G59S Tg and G71R Tg mice. (A-D) At 1, 6, 12, and 18 months of age, cohorts of male nTg, tTA, tetO-WT, and WT Tg mice (n = 10 per genotype) were repeatedly weighed (A); tested for ambulatory movement (B) and rearing (C) in the open-field test; and examined for the latency to fall in the rotarod test (D). (E-H) At 1, 6, 12, and 18 months of age, cohorts of male nTg (n = 10), tTA (n = 11), tetO-G59S (n = 10), and G59S Tg (n = 10) mice were repeatedly weighed (E); tested for ambulatory movement (F) and rearing (G) in the open-field test; and examined for the latency to fall in the rotarod test (H). (I-L) At 1, 6, 12, and 18 months of age, cohorts of male nTg (n = 11), tTA (n = 10), tetO-G71R (n = 10), and G71R Tg (n = 10) mice were repeatedly weighed (I); tested for ambulatory movement (J) and rearing (K) in the open-field test; and examined for the latency to fall in the rotarod test (L). Data are presented as mean ± SEM. Two-way ANOVA with Tukey’s multiple comparisons test was used for statistical analysis (at each time point, the mean of each genotype was compared with the mean of every other genotype). *P < 0.05, **P < 0.01, ****P < 0.0001.
G59S Tg and G71R Tg mice develop progressive degeneration of midbrain DA neurons
Since neuronal loss in substantia nigra is a pathological hallmark of Perry syndrome[9,10], the numbers of TH-positive midbrain DA neurons in the substantia nigra pars compacta (SNc) and ventral tegmental area (VTA) of WT Tg, G59S Tg, G71R Tg mice, and their control mice were counted using an unbiased stereological approach. WT Tg mice presented with no apparent loss of DA neurons in either the SNc or VTA at 1, 6, 12, and 24 months of age compared with control mice [Figure 3A-D]. G59S Tg mice displayed severe and early-onset degeneration of DA neurons in the midbrain, with approximately 13%, 28%, and 57% neuronal loss in the SNc at 6, 12, and 24 months of age, respectively, and 50% of neuronal loss in the VTA at 24 months of age [Figure 3A-D]. G71R Tg mice exhibited mild and late-onset degeneration of DA neurons in the midbrain, with approximately 25% neuronal loss in the SNc as well as the VTA at 24 months of age [Figure 3A-D]. Interestingly, besides neuronal loss, G59S Tg and G71R Tg mice showed remarked and progressive dystrophy of DA neurites in the substantia nigra pars reticulata (SNr) starting at two months of age [Figure 3A and E]. Therefore, expression of either G59S or G71R mutant p150Glued in midbrain DA neurons leads to substantial and progressive neurodegeneration.
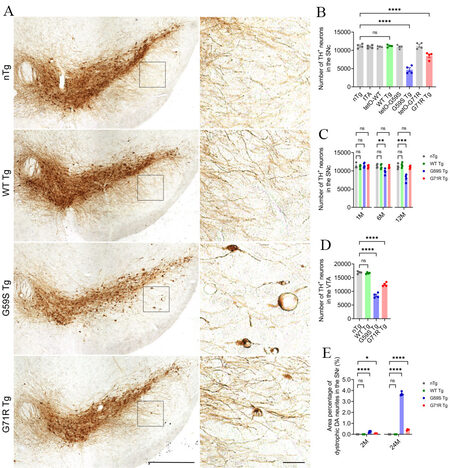
Figure 3. Progressive degeneration of DA neurons in the midbrain of G59S Tg and G71R Tg mice. (A) Immunohistochemical images show TH staining in the coronal midbrain sections of 24-month-old nTg, WT Tg, G59S Tg, and G71R Tg mice. Scale bar: (left) 500 μm (low-magnification images) and (right) 50 μm (high-magnification images). Note the dystrophic TH-positive DA neurites in the SNr of G59S Tg and G71R Tg mice. (B) Bar graphs depict the unbiased stereological estimation of the number of TH-positive DA neurons in the SNc of 24-month-old nTg, tTA, tetO-WT, WT Tg, tetO-G59S, G59S Tg, tetO-G71R, and G71R Tg mice (n = 4 per genotype). Data are presented as mean ± SEM. One-way ANOVA with Dunnett’s multiple comparisons test was used for statistical analysis (the mean of each genotype was compared with the mean of nTg). ****P < 0.0001. ns: Not significant. (C) Bar graphs depict the unbiased stereological estimation of the number of TH-positive DA neurons in the SNc of 1-, 6-, and 12-month-old nTg, WT Tg, G59S Tg, and G71R Tg mice (n = 4 per genotype per time point). Data are presented as mean ± SEM. At each time point, one-way ANOVA with Dunnett’s multiple comparisons test was used for statistical analysis (the mean of each genotype was compared with the mean of nTg). **P = 0.0099, ***P = 0.0001. (D) Bar graphs depict the unbiased stereological estimation of the number of TH-positive DA neurons in the VTA of 24-month-old nTg, WT Tg, G59S Tg, and G71R Tg mice (n = 4 per genotype). Data are presented as mean ± SEM. One-way ANOVA with Dunnett’s multiple comparisons test was used for statistical analysis (the mean of each genotype was compared with the mean of nTg). ****P < 0.0001. (E) Bar graphs quantify the area percentage of dystrophic TH-positive DA neurites in the SNr of 2- and 24-month-old nTg, WT Tg, G59S Tg, and G71R Tg mice (at each time point, n = 4 animals per genotype and 5 sections per animal). Dystrophic DA neurites were defined as neuritic varicosity ≥ 25 μm2. Data are presented as mean ± SEM. At each time point, one-way ANOVA with Dunnett’s multiple comparisons test was used for statistical analysis (the mean of each genotype was compared with the mean of nTg). *P = 0.0259, ****P < 0.0001. TH: Tyrosine hydroxylase.
G59S Tg and G71R Tg mice display robust degeneration of DA axon terminals
As axon dystrophy was reported in the striatum of patients with Perry syndrome[30,31], the density and morphology of DA axon terminals in the dorsal striatum of nTg, WT Tg, G59S Tg, and G71R Tg mice were examined. Compared with age-matched nTg controls, 2-, 12-, and 24-months old WT Tg mice had no apparent alteration of TH-positive DA axon terminals [Figure 4A and B]. However, G59S Tg and G71R Tg mice displayed substantial loss of DA axon terminals, with approximately 15%, 33%, and 55% of axonal loss in 6-, 12- and 24-month-old G59S Tg mice, respectively, and 35% of axonal loss in 24-month-old G71R Tg mice [Figure 4A and B]. Furthermore, TH staining also revealed abnormally enlarged DA axon terminals in the striatum of G59S Tg and G71R Tg mice [Figure 4A, arrow]. These data demonstrate that expression of either G59S or G71R mutant p150Glued in midbrain DA neurons causes robust degeneration of DA axon terminals and suggest dysfunctional DA transmission in G59S Tg and G71R Tg mice.
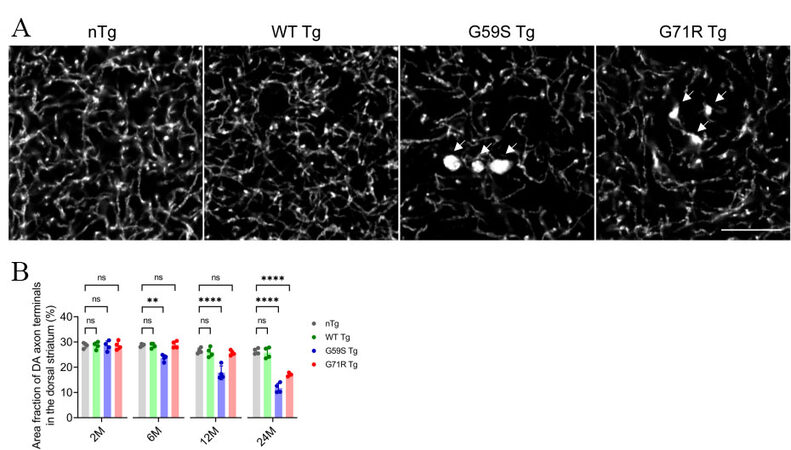
Figure 4. Robust degeneration of DA axon terminals in the striatum of G59S Tg and G71R Tg mice. (A) Representative images show TH staining in the dorsal striatum coronal sections of 24-month-old nTg, WT Tg, G59S Tg, and G71R Tg mice. Scale bar: 10 μm. Arrows point to swellings of TH-positive DA axon terminals in the striatum of G59S Tg and G71R Tg mice. (B) Bar graph estimates the area fraction of TH-positive DA axon terminals in the dorsal striatum of 2-, 6-, 12-, and 24-month-old nTg, WT Tg, G59S Tg, and G71R Tg mice (at each time point, n = 4 animals per genotype and 10 sections per animal). Data are presented as mean ± SEM. At each time point, one-way ANOVA with Dunnett’s multiple comparisons test was used for statistical analysis (the mean of each genotype was compared with the mean of nTg). **P = 0.0018, ****P < 0.0001. ns: Not significant. TH: Tyrosine hydroxylase.
G59S Tg and G71R Tg mice show alteration of dopamine transmission
Considering the pathological findings, the steady-state level of dopamine in the dorsal striatum of nTg, WT Tg, G59S Tg, and G71R Tg mice was assessed with HPLC. At 18 months of age, WT Tg and G71R Tg mice showed no apparent change in dopamine level compared with nTg controls, while G59S Tg mice displayed a significant reduction of dopamine content, with an approximately 53% of decrease [Figure 5A]. Regarding the primary dopamine metabolite 3,4-dihydroxyphenylacetic acid (DOPAC), neither its content nor the DOPAC/dopamine ratio in 18-month-old WT Tg, G59S Tg, and G71R Tg mice was significantly different with age-matched nTg controls [Figure 5A]. These data reveal dopamine deficiency in the striatum of G59S Tg mice, which might be the functional consequence of robust degeneration of midbrain DA neurons and the pathophysiological basis for the motor deficits observed in G59S Tg mice. By contrast, at 18 months of age, G71R Tg mice developed neither loss of midbrain DA neurons nor reduced dopamine content, further supporting the late-onset DA neuronal loss in G71R Tg mice.
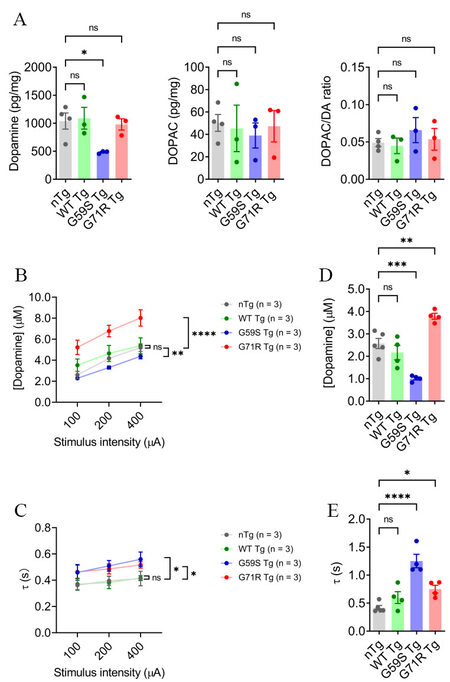
Figure 5. Alteration of dopamine content and release in the striatum of G59S Tg and G71R Tg mice. (A) HPLC measures the content of dopamine and DOPAC in the dorsal striatum of 18-month-old nTg, WT Tg, G59S Tg, and G71R Tg mice (n = 3 or 4 per genotype). Data are presented as mean ± SEM. One-way ANOVA with Dunnett’s multiple comparisons test was used for statistical analysis (the mean of each genotype was compared with the mean of nTg). *P = 0.0397. ns: Not significant. (B, C) FSCV quantifies the peak evoked dopamine release (B) and time constant of slope decay (C) following single-pulse electrical stimulation of different stimulus intensity in the dorsal striatum of six-month-old nTg, WT Tg, G59S Tg, and G71R Tg mice (n = 3 animals per genotype and 3 sections per animal). Data are presented as mean ± SEM. Two-way ANOVA genotype factor: nTg vs. G59S Tg, F(1,12) = 25.11, **P = 0.0044; nTg vs. G71R Tg, F(1,12) = 40.19, ****P < 0.0001 (B). Two-way ANOVA genotype factor: nTg vs. G59S Tg, F(1,12) = 9.285, *P = 0.0101; nTg vs. G71R Tg, F(1,12) = 8.491, *P = 0.0130 (C). (D,E) FSCV quantifies the peak evoked dopamine release (D) and time constant of slope decay (E) in response to burst electrical stimulation (200 μA, 50 Hz, 5 pulses) in the dorsal striatum of 18-month-old nTg, WT Tg, G59S Tg, and G71R Tg mice (n = 4 or 5 animals per genotype and 3 sections per animal). Data are presented as mean ± SEM. One-way ANOVA with Dunnett’s multiple comparisons test was used for statistical analysis (the mean of each genotype was compared with the mean of nTg). nTg vs. G59S Tg, ***P = 0.0004; nTg vs. G71R Tg, **P = 0.0034 (D). nTg vs. G59S Tg, ****P < 0.0001; nTg vs. G71R Tg, *P = 0.0357 (E).
Next, dopamine release kinetics in the dorsal striatum of nTg, WT Tg, G59S Tg, and G71R Tg mice was evaluated using fast-scan cyclic voltammetry (FSCV). At six months of age, WT Tg mice showed comparable evoked dopamine release and dopamine uptake in response to single-pulse stimulation compared with nTg controls [Figure 5B and C]. However, G59S Tg mice displayed significantly decreased dopamine release and slower dopamine uptake, while G71R Tg mice exhibited substantially increased dopamine release and slower dopamine uptake [Figure 5B and C]. Similarly, 18-month-old WT Tg mice showed comparable evoked dopamine release and dopamine uptake in response to burst-pulse stimulation compared with age-matched nTg controls [Figure 5D and E]. However, G59S Tg mice displayed significantly decreased dopamine release and slower dopamine uptake, while G71R Tg mice exhibited substantially increased dopamine release and slower dopamine uptake [Figure 5D and E]. Taken together, G59S Tg mice showed early-onset defects in striatal dopamine transmission, consistent with the early-onset loss of DA neurons and axons. In contrast, G71R Tg mice showed early-onset dysfunction of dopamine release and uptake long before the substantial loss of DA neurons and axons occurred. Thus, these data indicate that expression of either G59S or G71R mutant p150Glued in midbrain DA neurons profoundly compromises the dopamine transmission of the nigrostriatal DA pathway, contributing to the motor phenotypes developed in these transgenic mice.
G59S Tg and G71R Tg mice exhibit an age-dependent reduction of DA proteins
Tyrosine hydrolase (TH) is the rate-limiting enzyme in dopamine synthesis, and dopamine transporter (DAT) mediates dopamine uptake[32,33]. Since TH and DAT play critical roles in the nigrostriatal DA pathway, the protein levels of TH and DAT in the striatal homogenates of 1- and 24-month-old nTg, WT Tg, G59S Tg, and G71R Tg mice were examined. At one month of age, nTg, WT Tg, G59S Tg, and G71R Tg mice had comparable levels of TH and DAT in the striatum [Figure 6A and B]. At 24 months of age, WT Tg mice showed no marked change in TH and DAT protein expression compared with nTg controls, while both G59S Tg and G71R Tg mice displayed substantial loss of TH and DAT [Figure 6A and B]. Interestingly, the percentages of DAT loss in the striatum of 24-month-old G59S Tg and G71R Tg mice (approximately 70% and 42%, respectively) were more severe than the TH loss (about 53% and 29%, respectively) [Figure 6A and B]. These data suggest expression of either G59S or G71R mutant p150Glued in midbrain DA neurons may preferentially downregulate the expression and function of DAT protein at DA axon terminals.
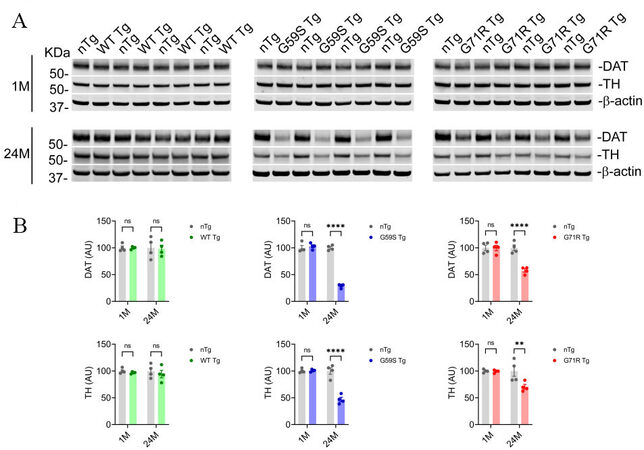
Figure 6. Reduction of dopaminergic proteins in the striatum of G59S Tg and G71R Tg mice. (A, B) Western blots show the expression of DAT and TH in the striatum homogenates of 1- and 24-month-old nTg, WT Tg, G59S Tg, and G71R Tg mice. Actin was used as the loading control. The bar graph estimates the levels of DAT and TH expression normalized against β-actin (n = 4 per genotype). Data are presented as mean ± SEM. Two-way ANOVA with Sidak’s multiple comparisons test was used for statistical analysis. **P = 0.0054, ****P < 0.0001. ns: Not significant.
G59S Tg mice display age-dependent accumulation of p150Glued aggregates in midbrain dopaminergic neurons
As both G59S and G71R mutant p150Glued were found aggregation-prone in vitro[17-19], the subcellular distribution pattern of p150Glued in the midbrain DA neurons of 2-, 6-, 12-, and 24-month-old nTg, WT Tg, G59S Tg, and G71R Tg mice was examined. TH and p150Glued co-staining revealed diffuse cytoplasmic localization of p150Glued without apparent aggregate formation in the soma and neurites of midbrain DA neurons from both young and aged nTg, WT Tg, and G71R Tg mice [Figure 7A and B]. By contrast, G59S Tg mice presented with p150Glued-positive cytoplasmic aggregates, mainly in the soma and in the neurites of midbrain DA neurons [Figure 7A]. Furthermore, the percentage of DA neurons with p150Glued aggregate in G59S Tg mice increased in an age-dependent manner, approximately 0%, 10.54%, 19.66%, and 82.17% at 2, 6, 12, and 24 months of age, respectively [Figure 7B]. Immunostaining further revealed the co-aggregation of dynactin subunit p50 and ARP1, as well as dynein subunit DLIC, with p150Glued aggregates inside the dystrophic DA neurites in the SNr of six-month-old G59S Tg mice [Figure 8A-C]. In addition, co-staining of coronal midbrain sections showed co-localization of lysosomes with p150Glued aggregates and a substantial increase of ubiquitin immunoreactivities within the dystrophic DA neurites in the SNr of six-month-old G59S Tg mice, compared with nTg controls [Figure 9A and B]. Thus, G59S mutation rather than G71R mutation induces mutant p150Glued to form cytoplasmic aggregates in midbrain DA neurons in vivo. Moreover, the aggregates trap dynein/dynactin, co-localize with lysosomes, and upregulate ubiquitination, implying detrimental effects of G59S mutant p150Glued on dynein/dynactin function and the degradative pathway in the midbrain DA neurons. These findings may explain why G59S Tg mice developed severe and early-onset degeneration of midbrain DA neurons, while G71R Tg mice showed milder and more slowly progressive neurodegeneration.
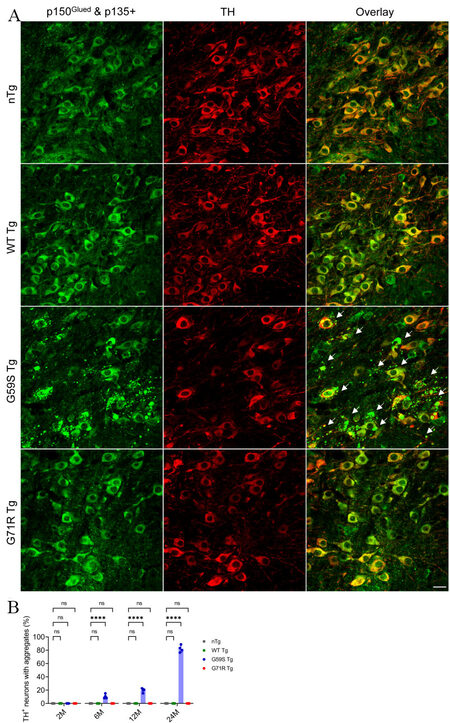
Figure 7. Formation of p150Glued-positive protein aggregates in midbrain DA neurons of G59S Tg mice. (A) Immunofluorescent images show the staining of p150Glued and p135+ (green) and TH (red) in midbrain coronal sections of 24-month-old nTg, WT Tg, G59S Tg, and G71R Tg mice. Scale bar: 20 μm. Arrows point to proteins aggregates of p150Glued and p135+ in the soma and neurites of DA neurons from G59S Tg mice. (B) Bar graph estimates the percentage of TH-positive neurons with proteins aggregates of p150Glued and p135+ in the midbrain of 2-, 6-, 12-, and 24-month-old nTg, WT Tg, G59S Tg, and G71R Tg mice (at each time point, n = 4 animals per genotype and 5 sections per animal). Aggregates were defined as puncta ≥ 1 μm2. Data are presented as mean ± SEM. At each time point, one-way ANOVA with Dunnett’s multiple comparisons test was used for statistical analysis (the mean of each genotype was compared with the mean of nTg). ****P < 0.0001. ns: Not significant. TH: Tyrosine hydroxylase.
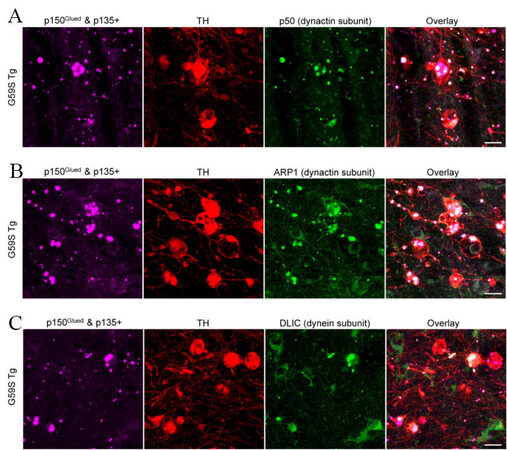
Figure 8. Co-aggregation of dynactin and dynein subunits with G59S mutation-induced p150Glued aggregates in the dystrophic DA neurites of G59S Tg mice. (A) Immunofluorescent images show the staining of p150Glued and p135+ (purple), TH (red), and dynactin subunit p50 (green) in the SNr of six-month-old G59S Tg mice. Scale bar: 20 μm. (B) Immunofluorescent images show the staining of p150Glued and p135+ (purple), TH (red), and dynactin subunit ARP1 (green) in the SNr of six-month-old G59S Tg mice. Scale bar: 20 μm. (C) Immunofluorescent images show the staining of p150Glued and p135+ (purple), TH (red), and dynein subunit DLIC (green) in the SNr of six-month-old G59S Tg mice. Scale bar: 20 μm.
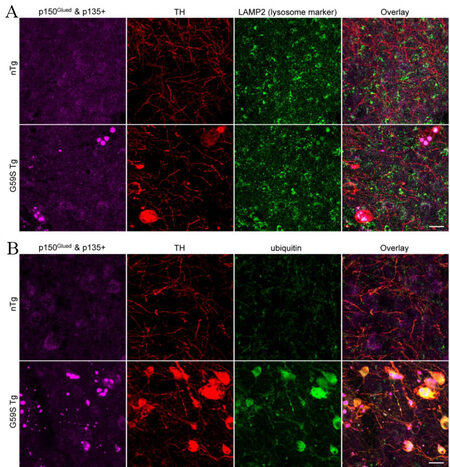
Figure 9. Co-localization of lysosomes with G59S mutation-induced p150Glued aggregates and upregulated ubiquitination in the dystrophic DA neurites of G59S Tg mice. (A) Immunofluorescent images show the staining of p150Glued and p135+ (purple), TH (red), and LAMP2 (green) in the SNr of six-month-old nTg and G59S Tg mice. Scale bar: 20 μm. (B) Immunofluorescent images show the staining of p150Glued and p135+ (purple), TH (red), and ubiquitin (green) in the SNr of six-month-old nTg and G59S Tg mice. Scale bar: 20 μm.
DISCUSSION
In this study, transgenic mouse lines with selective overexpression of wild-type human p150Glued, motor neuron disease-related G59S mutant p150Glued, or Perry syndrome-related G71R mutant p150Glued in midbrain DA neurons were generated and characterized. The WT Tg mice did not develop any overt motor phenotypes or midbrain DA neurodegeneration compared with age-matched nTg mice. The G59S Tg mice displayed weight loss, motor deficits, early-onset decreased dopamine transmission, and early-onset progressive loss of DA neurons and axons. The G71R Tg mice exhibited hyperactivities, impaired motor coordination, early-onset increased dopamine release and slower dopamine uptake, and late-onset loss of DA neurons and axons. Furthermore, both G59S Tg and G71R Tg mice showed an age-dependent reduction of DA proteins in the striatum, with a more severe loss of DAT than TH. Finally, G59S mutant p150Glued rather than G71R mutant p150Glued was found to form aggregates in mouse midbrain DA neurons in an age-dependent manner, and the aggregates trapped dynein/dynactin, co-localized with lysosomes, and upregulated ubiquitination.
Since the initial identification of motor neuron disease- and Perry syndrome-causative mutations in p150Glued, several mouse strains of mutant p150Glued have been generated to study their effects on selective degeneration of specific types of neurons. Motor neuron disease-like motor phenotypes and degeneration of spinal motor neurons have been reported in G59S p150Glued transgenic and knock-in mouse models[20-22]. Decreased exploratory activity and impaired motor coordination were observed in the transgenic mice overexpressing G71A p150Glued under the control of the Thy1 promotor[23]. However, either the expression of G71A p150Glued in midbrain DA neurons or neuronal loss in the brain tissue was detected in the transgenic mice[23]. Recently, G71A p150Glued knock-in mouse models were created[24]. Increased immobility time in the tail suspension test, impaired motor coordination in the beam-walking test, and decreased TH staining intensity in the substantia nigra neurons were found in the heterozygous G71A p150Glued knock-in mice[24], recapitulating some of the clinical features of Perry syndrome. However, the pathogenic effects of mutant p150Glued on Perry syndrome-related degeneration of the nigrostriatal DA pathway remain to be determined. New lines of transgenic mice (WT Tg, G59S Tg, and G71R Tg) with selective overexpression of wild-type, G59S mutant, or G71R mutant human p150Glued in midbrain DA neurons were generated in the current study, allowing us to systematically evaluate the impact of mutant p150Glued on survival and function of midbrain DA neurons. WT Tg mice showed neither motor phenotypes nor pathological, functional, or biochemical abnormalities of the nigrostriatal DA pathway. G59S Tg mice displayed substantial weight loss, dramatic reduction of rearing movement in the open field test, and significant impairment of motor coordination in the rotarod test. The pathophysiological basis for the motor phenotypes in G59S Tg mice is the early-onset and severe loss of DA neurons and axon terminals and the resultant deficiency of dopamine content and transmission. G71R Tg mice exhibited significant hyperactivities in ambulatory and rearing movements and substantially impaired rotarod performance through 6-18 months of age. In contrast to the severe and early-onset degeneration of nigrostriatal DA pathway in G59S Tg mice, the neurodegeneration in G71R mice was much milder and more slowly progressive, with no significant loss of DA neurons and axon terminals occurring before 18 months of age. Despite no early-onset DA neuron degeneration, G71R Tg mice showed an early-onset increase of dopamine release and slower dopamine uptake in the striatum, which may elevate the extracellular dopamine level and contribute to hyper-dopamine motor phenotypes. Thus, our data demonstrate that both G59S and G71R mutant p150Glued are pathogenic to the nigrostriatal DA pathway in vivo, but differentially impair the survival and functions of midbrain DA neurons. The finding on G59S mutation is unexpected since the mutation was particularly linked to a slowly progressive autosomal dominant form of lower motor neuron disease, the distal hereditary motor neuropathy 7B (HMN7B)[6]. Our previous study showed that both homozygous Dctn1 knock-out mice and homozygous DCTN1 G59S knock-in mice died during early embryogenesis, indicating the loss-of-function nature of G59S mutation[20]. Furthermore, we found that the G59S mutation disrupts the stability of p150Glued protein, so we could not detect any expression and accumulation of p150Glued protein in the tissues of homozygous DCTN1 G59S knock-in embryos[20]. By contrast, in the G59S Tg mice, an artificial overexpression of G59S mutation in DA neurons led to an extensive accumulation of misfolded p150Glued proteins, which may overwhelm the protein degradation system in the DA neurons and eventually induce cell death. Therefore, the overexpression data in either cell or animal models must be interpreted with caution.
Microtubule-binding domains in p150Glued are required for the initiation of retrograde axonal transport, stability of axonal microtubules, and integrity of axon terminals[13-16]. Accordingly, degeneration of ventral root axons and disruption of neuromuscular junctions have been observed in G59S mutant p150Glued transgenic mice[21,22]. In line with these earlier findings, the current study revealed that expression of either G59S or G71R mutant p150Glued in midbrain DA neurons resulted in substantial dysfunction and degeneration of DA axon terminals in the striatum. Additionally, our study showed expression of either G59S or G71R mutant p150Glued in midbrain DA neurons led to the dystrophy of DA neurites in the SNr, indicating an essential role of p150Glued in maintaining the structural integrity of both axons and dendrites of DA neurons. Future studies will be needed to determine the underlying mechanism of how mutant p150Glued triggers the structural and functional changes of distal and proximal DA neurites.
Several mechanisms have been hypothesized to address the selective degeneration of motor neurons caused by G59S mutant p150Glued, including impaired retrograde axonal transport of trophic signaling, defects of vesicular trafficking in the ER-Golgi secretory pathway, aberrant autophagosome-lysosome degradative pathway, and cytotoxicity of mutant p150Glued aggregates[17,21,22]. However, due to the lack of appropriate animal models, mechanisms for the selective degeneration of DA neurons in Perry syndrome have been poorly understood. Until recently, DCTN1 G71A knock-in mice were generated, and homozygous knock-in mice died prenatally, suggesting a potential loss-of-function nature of the mutation[24]. In the heterozygous DCTN1 G71A knock-in mice, a reduction of TH immunoreactivity in DA neurons was reported[24]. However, the effect of mutant p150Glued on dopamine transmission and the related regulating proteins remains to be determined. Our HPLC and FSCV studies revealed that overexpression of human G59S p150Glued in midbrain DA neurons led to substantial dopamine deficiency, significantly decreased dopamine release, and slower dopamine uptake, reflecting the severe and early-onset loss of DA neurons and axons. In contrast, overexpression of human G71R p150Glued in midbrain DA neurons resulted in early-onset substantially increased dopamine release and slower dopamine uptake long before the loss of DA axons and the deficiency of dopamine content in the striatum occurred, indicating the early-onset dysfunction of DAT. Correlatively, Western blot analysis demonstrated that overexpression of either G59S or G71R mutant p150Glued in midbrain DA neurons preferentially downregulated the expression of DAT at DA axon terminals. Further investigation will be required to unmask the specific cellular mechanisms leading to the DAT dysfunction and the selective cell death of DA neurons in Perry syndrome.
G59S mutation, as well as all the Perry syndrome-causative mutations, occurs within or close to the CAP-Gly domain of p150Glued, causing loss of function of the MTBDs[6-8]. G59S mutant p150Glued is prone to misfold and form cytoplasmic aggregates leading to enhanced cell death both in vitro and in vivo, suggesting both loss-of-function and toxic-gain-of-function mechanisms are involved in G59S mutation-induced neurodegeneration[17,21]. Perry syndrome-related G71A mutant p150Glued has been reported to aggregate less than G59S mutant p150Glued when overexpressed in HeLa cells[18]. Moreover, no evident p150Glued aggregates are observed in G71A transgenic mice[23]. In line with these earlier findings, our study revealed that neither wild-type nor G71R mutant p150Glued formed aggregates in vivo when overexpressed in midbrain DA neurons. In contrast, G59S mutant p150Glued formed aggregates in an age-dependent way when overexpressed in mouse midbrain DA neurons. Moreover, co-aggregation of dynactin and dynein subunits with G59S mutation-induced p150Glued aggregates was observed inside the dystrophic DA neurites of G59S Tg mice. The data suggest the sequestration of active motors in aggregates; the restricted accessibility of adaptors, cargoes, and regulatory proteins to dynein/dynactin; and thus the impairment of normal functions of dynein/dynactin in the midbrain DA neurons. In addition, co-localization of lysosomes with G59S mutation-induced p150Glued aggregates and upregulated ubiquitination were found within the dystrophic DA neurites of G59S Tg mice, implying the disruption of the degradative pathway in the midbrain DA neurons. These observations may explain why the degeneration of the nigrostriatal DA pathway in G59S Tg mice is more early-onset and severe than in G71R Tg mice in our study. Future studies will explore whether overexpression of exogenous G59S or G71R mutant p150Glued compromises the normal functions of endogenous dynactin p150Glued in the midbrain DA neurons to test if there is a dominant-negative mechanism for DCTN1 mutation-induced degeneration of nigrostriatal DA pathway.
While the present study focused on the role of mutant p150Glued in Perry syndrome-related parkinsonism and the underlying degeneration of midbrain DA neurons, the pathophysiological basis and pathogenic mechanisms of other Perry syndrome-related symptoms remain to be delineated in detail, including apathy/depression and central hypoventilation[9,10]. Except for the loss of midbrain DA neurons, Perry syndrome patients also display marked degeneration of norepinephrine neurons in locus coeruleus and serotonergic neurons in the medullary raphe and ventrolateral medulla[34,35]. Future studies will be needed to crossbreed our tetO-human DCTN1 with suitable tTA lines and determine the pathogenic significance of mutant p150Glued on norepinephrine neurons and serotonergic neurons.
In summary, our findings show selective expression of either G59S or G71R mutant p150Glued in mouse midbrain DA neurons leads to parkinsonism-like motor abnormalities and degeneration of the nigrostriatal DA pathway, recapitulating some key clinical and pathological features of Perry syndrome. Moreover, G59S and G71R mutant p150Glued exhibit differential pathogenic impact on the survival and function of midbrain DA neurons in vivo. The p150Glued transgenic moue models provide valuable tools for mechanistic insights and therapeutic targets of DA neuron degeneration in Perry syndrome.
DECLARATIONS
AcknowledgmentsThe authors thank Dr. David M Lovinger of National Institute on Alcohol Abuse and Alcoholism for assisting the FSCV recording, members of Cai and Yu Labs for various supports, and Beijing Hospital Authority for the Science & Technology Innovation Program and High-level Talents Program.
Authors’ contributionsDesigned the study, analyzed the data, and wrote the manuscript: Cai H, Yu J, Sgobio C
Performed the experiments: Yu J, Sgobio C, Yang X, Peng Y, Chen X, Sun L, Shim H
Read and approved the final version of the manuscript: all authors
Availability of data and materialsData and materials of the current study will be available upon reasonable request.
Financial support and sponsorshipThis work was supported by the intramural research programs of National Institute on Aging (AG000946), National Natural Science Foundation of China (No. 82071438 and 81601117), Beijing Natural Science Foundation (No. 7202077), Beijing BaiQianWan Talents Project (No. 2017002), and Beijing Nova Program (No. xx2018099).
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateAll animal procedures conformed to the NIH (13-040) guide for the ethical care and use of laboratory animals. Animal protocols were approved by the Institutional Animal Care and Use Committee of National Institute on Aging and Beijing Geriatric Hospital.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. Reck-Peterson SL, Redwine WB, Vale RD, Carter AP. The cytoplasmic dynein transport machinery and its many cargoes. Nat Rev Mol Cell Biol 2018;19:382-98.
2. Cianfrocco MA, DeSantis ME, Leschziner AE, Reck-Peterson SL. Mechanism and regulation of cytoplasmic dynein. Annu Rev Cell Dev Biol 2015;31:83-108.
3. Bercier V, Hubbard JM, Fidelin K, et al. Dynactin1 depletion leads to neuromuscular synapse instability and functional abnormalities. Mol Neurodegener 2019;14:27.
5. Konno T, Ross OA, Teive HAG, Sławek J, Dickson DW, Wszolek ZK. DCTN1-related neurodegeneration: Perry syndrome and beyond. Parkinsonism Relat Disord 2017;41:14-24.
6. Puls I, Jonnakuty C, LaMonte BH, et al. Mutant dynactin in motor neuron disease. Nat Genet 2003;33:455-6.
7. Farrer MJ, Hulihan MM, Kachergus JM, et al. DCTN1 mutations in Perry syndrome. Nat Genet 2009;41:163-5.
8. Zhang J, Wang H, Liu W, et al. A novel Q93H missense mutation in DCTN1 caused distal hereditary motor neuropathy type 7B and Perry syndrome from a Chinese family. Neurol Sci 2021;42:3695-705.
9. Dulski J, Cerquera-Cleves C, Milanowski L, et al. Clinical, pathological and genetic characteristics of Perry disease-new cases and literature review. Eur J Neurol 2021;28:4010-21.
10. Tsuboi Y, Mishima T, Fujioka S. Perry disease: concept of a new disease and clinical diagnostic criteria. J Mov Disord 2021;14:1-9.
11. Dixit R, Levy JR, Tokito M, Ligon LA, Holzbaur EL. Regulation of dynactin through the differential expression of p150Glued isoforms. J Biol Chem 2008;283:33611-9.
12. Zhapparova ON, Bryantseva SA, Dergunova LV, et al. Dynactin subunit p150Glued isoforms notable for differential interaction with microtubules. Traffic 2009;10:1635-46.
13. Lloyd TE, Machamer J, O’Hara K, et al. The p150(Glued) CAP-Gly domain regulates initiation of retrograde transport at synaptic termini. Neuron 2012;74:344-60.
14. Moughamian AJ, Holzbaur EL. Dynactin is required for transport initiation from the distal axon. Neuron 2012;74:331-43.
15. Lazarus JE, Moughamian AJ, Tokito MK, Holzbaur EL. Dynactin subunit p150(Glued) is a neuron-specific anti-catastrophe factor. PLoS Biol 2013;11:e1001611.
16. Yu J, Lai C, Shim H, et al. Genetic ablation of dynactin p150Glued in postnatal neurons causes preferential degeneration of spinal motor neurons in aged mice. Mol Neurodegener 2018;13:10.
17. Levy JR, Sumner CJ, Caviston JP, et al. A motor neuron disease-associated mutation in p150Glued perturbs dynactin function and induces protein aggregation. J Cell Biol 2006;172:733-45.
18. Ishikawa K, Saiki S, Furuya N, et al. P150glued-associated disorders are caused by activation of intrinsic apoptotic pathway. PLoS One 2014;9:e94645.
19. Mishima T, Ishikawa T, Imamura K, et al. Cytoplasmic aggregates of dynactin in iPSC-derived tyrosine hydroxylase-positive neurons from a patient with Perry syndrome. Parkinsonism Relat Disord 2016;30:67-72.
20. Lai C, Lin X, Chandran J, Shim H, Yang WJ, Cai H. The G59S mutation in p150(glued) causes dysfunction of dynactin in mice. J Neurosci 2007;27:13982-90.
21. Laird FM, Farah MH, Ackerley S, et al. Motor neuron disease occurring in a mutant dynactin mouse model is characterized by defects in vesicular trafficking. J Neurosci 2008;28:1997-2005.
22. Chevalier-Larsen ES, Wallace KE, Pennise CR, Holzbaur EL. Lysosomal proliferation and distal degeneration in motor neurons expressing the G59S mutation in the p150Glued subunit of dynactin. Hum Mol Genet 2008;17:1946-55.
23. Mishima T, Deshimaru M, Watanabe T, et al. Behavioral defects in a DCTN1G71A transgenic mouse model of Perry syndrome. Neurosci Lett 2018;666:98-103.
24. Deshimaru M, Mishima T, Watanabe T, et al. Behavioral profile in a Dctn1G71A knock-in mouse model of Perry disease. Neurosci Lett 2021;764:136234.
25. Wang L, Xie C, Greggio E, et al. The chaperone activity of heat shock protein 90 is critical for maintaining the stability of leucine-rich repeat kinase 2. J Neurosci 2008;28:3384-91.
26. Lin X, Parisiadou L, Gu XL, et al. Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson’s-disease-related mutant alpha-synuclein. Neuron 2009;64:807-27.
27. Schmoldt A, Benthe HF, Haberland G. Digitoxin metabolism by rat liver microsomes. Biochem Pharmacol 1975;24:1639-41.
28. Liu G, Sgobio C, Gu X, et al. Selective expression of Parkinson’s disease-related Leucine-rich repeat kinase 2 G2019S missense mutation in midbrain dopaminergic neurons impairs dopamine release and dopaminergic gene expression. Hum Mol Genet 2015;24:5299-312.
29. Yorgason JT, España RA, Jones SR. Demon voltammetry and analysis software: analysis of cocaine-induced alterations in dopamine signaling using multiple kinetic measures. J Neurosci Methods 2011;202:158-64.
30. Felicio AC, Dinelle K, Agarwal PA, et al. In vivo dopaminergic and serotonergic dysfunction in DCTN1 gene mutation carriers. Mov Disord 2014;29:1197-201.
31. Chung EJ, Hwang JH, Lee MJ, et al. Expansion of the clinicopathological and mutational spectrum of Perry syndrome. Parkinsonism Relat Disord 2014;20:388-93.
32. Nagatsu T, Nakashima A, Ichinose H, Kobayashi K. Human tyrosine hydroxylase in Parkinson’s disease and in related disorders. J Neural Transm (Vienna) 2019;126:397-409.
33. Mulvihill KG. Presynaptic regulation of dopamine release: role of the DAT and VMAT2 transporters. Neurochem Int 2019;122:94-105.
34. Wider C, Dickson DW, Stoessl AJ, et al. Pallidonigral TDP-43 pathology in Perry syndrome. Parkinsonism Relat Disord 2009;15:281-6.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Yu J, Sgobio C, Yang X, Peng Y, Chen X, Sun L, Shim H, Cai H. Selective expression of neurodegenerative diseases-related mutant p150Glued in midbrain dopaminergic neurons causes progressive degeneration of nigrostriatal pathway. Ageing Neur Dis 2022;2:6. http://dx.doi.org/10.20517/and.2022.07
AMA Style
Yu J, Sgobio C, Yang X, Peng Y, Chen X, Sun L, Shim H, Cai H. Selective expression of neurodegenerative diseases-related mutant p150Glued in midbrain dopaminergic neurons causes progressive degeneration of nigrostriatal pathway. Ageing and Neurodegenerative Diseases. 2022; 2(2): 6. http://dx.doi.org/10.20517/and.2022.07
Chicago/Turabian Style
Yu, Jia, Carmelo Sgobio, Xuan Yang, Yu Peng, Xi Chen, Lixin Sun, Hoon Shim, Huaibin Cai. 2022. "Selective expression of neurodegenerative diseases-related mutant p150Glued in midbrain dopaminergic neurons causes progressive degeneration of nigrostriatal pathway" Ageing and Neurodegenerative Diseases. 2, no.2: 6. http://dx.doi.org/10.20517/and.2022.07
ACS Style
Yu, J.; Sgobio C.; Yang X.; Peng Y.; Chen X.; Sun L.; Shim H.; Cai H. Selective expression of neurodegenerative diseases-related mutant p150Glued in midbrain dopaminergic neurons causes progressive degeneration of nigrostriatal pathway. Ageing. Neur. Dis. 2022, 2, 6. http://dx.doi.org/10.20517/and.2022.07
About This Article
Copyright

Data & Comments
Data


 Cite This Article 8 clicks
Cite This Article 8 clicks

 Like This Article 25
likes
Like This Article 25
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.