
Newly identified transmembrane protein 106B amyloid fibrils in the human brain: pathogens or by-products?
 0
0Abstract
Neurodegenerative diseases (NDs) such as Alzheimer’s disease (AD) and Parkinson’s disease (PD) constitute a spectrum of diseases characterized by the abnormal aggregation of specific amyloid fibrillar proteins; these include β-amyloid (Aβ) and tau in the form of the extracellular Aβ plaques and neuronal neurofibrillary tangles in AD and fibrillar α-synuclein aggregation in the form of Lewy bodies and Lewy neurites in PD. Transmembrane protein 106B (TMEM106B) is a type II transmembrane lysosomal protein that participates in lysosome morphology, localization, acidification, and trafficking; it is involved in the pathogenesis of several NDs, especially frontotemporal lobular degeneration with TAR DNA-binding protein immunoreactive inclusions (FTLD-TDP). Studies from four independent research groups revealed that the luminal domain of TMEM106B (120-254aa) forms amyloid fibrils in several brain regions in patients with a series of NDs and neurologically normal older adults. Given its potentially critical roles in the pathogenesis of NDs and brain aging, this surprising finding has focused attention on TMEM106B and suggested that it is nearly as fundamental as other pathogenic amyloid proteins (e.g., Aβ, tau,
Keywords
INTRODUCTION
Neurodegenerative diseases (NDs) are diverse and characterized by the abnormal deposition and aggregation of specific fibrillar proteins; they are thus classified by their aggregated proteins. NDs mainly consist of tauopathies, α-synucleinopathies and TAR DNA-binding protein (TDP-43) proteinopathies. The tauopathies mainly include Alzheimer’s disease (AD), progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), argyrophilic grain disease (AGD), and Pick’s disease (PiD). α-Synucleinopathies include Parkinson’s disease (PD), multiple system atrophy (MSA), and dementia with Lewy bodies (DLB). TDP-43 proteinopathies include amyotrophic lateral sclerosis (ALS) and frontotemporal lobular dementia (FTLD)[1,2]. Patients with different proteinopathies show distinct clinical features in personality, cognition, behavior, language, and movement. Moreover, even in patients with the same proteinopathies, there is remarkable heterogeneity in clinical and pathological manifestations. For example, PD (the most common α-synucleinopathy and movement disorder) presents with bradykinesia, resting tremor, and rigidity and is pathologically characterized by the α-syn amyloid aggregation in the form of Lewy bodies (LBs) and Lewy neurites (LNs). While MSA presents as various combinations of autonomic failure, cerebellar ataxia, and parkinsonism, and its pathological hallmark is the accumulation of α-syn in oligodendrocytes as glial cytoplasmic inclusions[3]. Several lines of evidence suggest that amyloid fibrillar proteins accumulating in NDs possess prion-like seeding and propagation properties and show various biological conformations[1,4-6]; these drive the initiation and progression of NDs. In recent years, the atomic structure of the amyloid fibrils extracted from brain samples of patients with NDs was determined by cryo-electronic microscopy (cryo-EM). The results suggest that not only the amyloid fibrils composed of different proteins but also those formed by the same protein but from different NDs show distinct conformations[7-15], which further support the hypothesis of “one strain, one disease” in NDs. The elucidation of the structure of pathogenic amyloid fibrils sheds new light on the pathomechanisms and therapeutic strategies for NDs.
A genome-wide association study (GWAS) showed that variants of transmembrane protein 106B
In addition to other pathogenic amyloid proteins in NDs, several research groups worldwide reported that TMEM106B forms amyloid fibril in the brains of NDs patients and neurologically normal older adults. Furthermore, the atomic structures of TMEM106B fibrils were determined using cryo-EM[29-32], which updated our view on the involvement of TMEM106B in the pathogenesis of NDs. Therefore, this review aims to introduce the physiological function of TMEM106B and summarize the findings of research that focus on exploring the mechanism and function of TMEM106B in NDs occurrence and progression. Next, we elucidate the identification of the TMEM106B fibril and its cryo-EM structures. Finally, we outline directions for future research and raise questions needed to be addressed about TMEM106B in NDs based on the implications produced by the identification of TMEM106B amyloid fibril in the human brain.
STRUCTURE OF TMEM106B IN THE NATIVE STATE
TMEM106B is a single-pass type II membrane protein that localizes in late endosomes/lysosomes[26,27,33]. TMEM106B is expressed in many tissues and organs (https://www.ncbi.nlm.nih.gov/gene/54664), especially in neurons and oligodendrocytes in the central nervous system[26,34-36]. It consists of 274 amino acids and is divided into an N-terminal domain (NTD, 1-96 aa), a transmembrane domain (TMD, 97-117aa), and a C-terminal domain (CTD, 118-274 aa). The NTD is intrinsically disordered and locates in the cytosol[37], following the TMD across the phospholipid bilayer of late endosome/lysosome once in the form of an α-helix; the CTD in the lumen is predicted to form seven β-sheets and is prone to aggregating[31,33]. Like other membrane proteins, TMEM106B is a highly glycosylated protein with N-glycosylation occurring at Asn 145, 151, 164, 183, and 254 in the CTD[33]. Two types of glycosylation with different functions occur in TMEM106B. Noncomplex glycosylation at N145, N151, and N164 does not influence TMEM106B localization. By contrast, the deficiency of complex glycosylation at N183 disrupts the normal transportation of TMEM106B to late endosomes/lysosomes. It leads to the accumulation of TMEM106B in the endoplasmic reticulum, suggesting that N183 glycosylation is required for the anterograde trafficking of TMEM106B to late endosomes/lysosomes. The other complex glycosylation at N254 appears to directly affect the sorting of TMEM106B to endosome based on the observation that the mutant of N254 resulted in significant localization of TMEM106B to the cell surface[33,38,39].
THE PHYSIOLOGICAL FUNCTION OF TMEM106B
Little is known about the function and cellular and molecular mechanisms of TMEM106B under physiological conditions; studies suggest that TMEM106B exerts its effects on the lysosome. Overexpression of TMEM10B in neuronal cell lines resulted in decreased numbers of lysosomes in cells and increased volume of lysosomes compared to the control group[26,34]. The enlarged lysosomes were also observed in the Oli-Neu oligodendrocyte cell line when TMEM106B was overexpressed[35]. The knockdown of TMEM106B using siRNA led to an approximately 50%-70% decrease in TMEM106B expression and did not influence lysosome number and morphology[26,27]. Two possibilities may explain these unapparelled findings. One is that the residual expression of TMEM106B after it is treated with siRNA is sufficient to maintain its normal function on lysosomal number and morphology. The other is that loss of function does not influence the - TMEM106B morphology or function. However, there was a conflicting result in which the numbers of lysosomes were significantly reduced when TMEM106B was knocked out using CRISPR/Cas-9 in an oligodendrocyte cell line[35], suggesting potentially varying effects of TMEM106B on lysosome numbers across cell types.
In addition to influencing the morphology and numbers of lysosomes, TMEM106B modulates the positioning and trafficking of lysosome vesicles. TMEM106B knockdown or knockout altered the localization of lysosomes and led to the formation of clusters because of the increased number of lysosomes near the nucleus in neurons, oligodendrocytes, and fibroblasts[27,35] but did not alter cell viability[27]. Under TMEM106B knockdown in primary neurons, the dendritic branching (mainly distal branches) decreased, and the retrograde trafficking of lysosomes along dendrites increased, whereas the number of lysosomes taking part in anterograde trafficking was unaffected. The imbalance between anterograde and retrograde trafficking vesicles may cause reduced branch complexity.
TMEM106B interacts with microtubule-associated protein 6 (MAP6) in the brain through its disordered NTD binding to the CTD of MAP6. Moreover, the overexpression of MAP6 in primary neurons mimicked the phenotypes of TMEM106B knockdown, and the knockdown of MAP6 rescued the dendritic arborization and the retrograde trafficking of lysosome[27]. These findings suggest that TMEM106B takes control of the dendritic lysosomal transport through its interaction with MAP6, which is critical in dendrite branching and maintenance.
Intraluminal pH is critical for maintaining normal lysosome function under physiological conditions[40,41], and TMEM106B regulates lysosome pH. Chen-Plotkin et al. reported that the LAMP1-positive organelles in HEK293 and Hela cells with TMEM106B overexpression were not as acidic as the control group with normal TMEM106B expression level[34], which leads to functional impairment of protein degradation. However, a study found that HEK293 cells transfected with wild-type TMEM106B presented more robust acidification than those control cells[35]. Furthermore, another study found that the deficiency of TMEM106B in primary neurons significantly impaired lysosomal acidification[28]. Vacuolar-ATPase (V-ATPase) is responsible for normal lysosomal acidification. The co-immunoprecipitation assay identified that TMEM106B interacts with accessory protein 1 (AP1, a subunit of V-ATPase) under physiological conditions. The deficiency of TMEM106B contributes to AP1 downregulation, disturbs the normal function of V-ATPase, and thus leads to lysosomal acidifying dysfunction[28]. In brief, TMEM10B plays essential roles in lysosome morphology, intracellular localization, trafficking, and acidification.
THE INVOLVEMENT OF TMEM106B IN NDS AND AGING
Although TMEM106B was initially identified as a risk factor for FTLD[16], studies revealed that TMEM106B variants are associated with varying clinical and pathological phenotypes of several NDs. Six SNPs of TMEM106B are thought to be associated with NDs, and five out of these are located in the non-coding regions of TMEM106B and do not result in the mutations of the TMEM106B protein. Nevertheless, these SNPs may regulate the expression of TMEM106B in NDs by influencing the alternative splicing of TMEM106B mRNA. Only one of the six SNPs, rs3173615, is located in the coding regions of TMEM106B and contributes to the nonsynonymous mutation p.T185S. The relationships between SNPs of TMEM106B and NDs are summarized in Table 1. TMEM106B is also involved in aging, one of the most potent risk factors for NDs[42].
Summaries of clinical/GWAS studies about SNPs of TMEM106B and its relationships with NDs
| NDs | TMEM106B SNPs | Cohort composition | Major allele | Minor allele | Relationships |
| FTLD | rs1990622 | FTLD-TDP, n = 515; control, n = 2509 | T | C | Major allele increases the risk of developing FTLD[16] |
| FTLD, n = 179; control, n = 137 | T | C | TC heterozygous carriers in FTLD show a more rapid decrease in cognitive function[20] | ||
| FTLD-GRN+, n = 27; FTLD-GRN-, n = 23; control, n = 73 | A | G | Individuals carrying the major allele have an earlier age at onset[53] | ||
| FTLD, n = 297; control, n = 595 | T | C | Minor allele reduces the risk of developing FTLD[55 | ||
| Discovery cohort: FTLD-C9orf72+, n = 14 Replicate cohort: FTLD-C9orf72+, n = 75 | T | C | The major allele is associated with later age at onset and death in C9orf72 expansion carriers[17] | ||
| GRN+, n = 17; control, n = 14 | T | C | In GRN+ individuals, the major allele is associated with decreased connectivity within the ventral salience network and the left frontoparietal network[99] | ||
| C9orf72+, n = 33; GRN+, n = 61; MAPT+, n = 14; control, n = 123 | C | T | In mutation carriers, minor allele enhances the benefit of cognitive reserve and modulates the slope of the correlation between education and grey matter volume[100] | ||
| FTLD/ALS-TDP, n = 90 | A | G | Individuals with AA genotype have a higher risk of neuro-astroglial tauopathy[101] | ||
| rs3173615 | Cohort 1: FTLD- C9orf72+, n = 325; cohort 2: FTLD- C9orf72+, n = 586; control, n = 1302 | G | C | Minor allele protects C9orf72 expansion carriers from developing FTLD[59] | |
| Discovery cohort: GRN+, n = 382; control, n = 1146; replicate cohort: GRN+, n = 210; control, n = 1798 | C | G | The minor allele is associated with lower odds of developing disease symptoms in GRN mutation carriers[19] | ||
| AD | rs1990622 | AD, n = 907 | T | C | Minor allele decreases the risk of developing hippocampal sclerosis[65] |
| LOAD, n = 1133; control, n = 1159 | T | C | In APOE e4 allele carriers, the minor allele has a higher frequency in AD than those without AD[63] | ||
| Typical AD, n = 807; LP-AD, n = 151; HpScl-AD, n = 132; HpScl, n = 30 | T | C | The major allele has a higher frequency in the HpScl and HpScl-AD than the typical AD and LP-AD[102] | ||
| AD, n = 21982; control, n = 41944; | T | C | Major allele increases the risk of developing AD[21] | ||
| rs1595014 | Discovery cohort: AD, n = 17536; control, n = 36175; replicate cohort:AD, n = 13219; Control, n = 4116; | A | T | AD risk is significantly influenced by the interaction of APOE with rs1595014 in TMEM106B[64] | |
| rs1548884 | Discovery cohort: AD, n = 154; MCI, n = 401; Control, n = 122; replicate cohort: AD, n = 70; MCI, n = 151; control, n = 87 | C | A | Rs1548884 is associated with CSF NFL level in all individuals involved[103] | |
| ALS | rs1990622 | ALS, n = 85; control, n = 553 | T | C | The major allele is associated with poor cognitive function[67] |
| ALS, n = 110 | T | C | Minor allele increases TDP-43 pathology[22] | ||
| ALS, n = 865 | A | G | The major allele is associated with a higher frequency of bulbar site of onset. Minor allele increases cognitive impairment[69] | ||
| PD | rs1990622 | PD, n = 179; control, n = 137 | T | C | PD patients carrying major alleles present a faster decline of cognitive function over time[20] |
| PD, n = 1121; control, n = 829 | C | T | Minor allele increases the risk for PD patients with initial symptom of rigidity/bradykinesia[104] | ||
| HS-aging | rs1990622 | HS-Aging, n = 268; control, n = 2957 | A | G | AA genotypes carriers have a higher risk for developing HS-Aging pathology[74] |
TMEM106B in FTLD
FTLD is a group of heterogeneous and devastating neurodegenerative syndromes and is the third leading cause of dementia after AD and DLB[43]. Pathologically, it is characterized by aggressive atrophy in the frontal and temporal lobes, contributing to corresponding clinical manifestations, including progressive behavioral deficits, personality alterations, executive dysfunction, and impaired speech[44]. FTLD shows the most significant pathological variability among NDs. The predominant neuropathology in FTLD is TDP-43 immunoreactive inclusions in neurons (FTLD-TDP), accounting for about 50% of all FTLDs[45]. Moreover, FTLD-TDP is classified into five subscales (Types A to E) based on the morphology of neuronal TDP-43 immuno-positive inclusions and the affected neocortical layers[46,47]. Although most FTLD is sporadic, approximately 10%-20% of FTLD is hereditary in an autosomal dominant manner[48]. The common pathogenic genes in FTLD include GRN, microtubule-associated protein tau
A GWAS identified susceptibility loci for FTLD-TDP and found that three SNPs (rs1990622, rs6966915, and rs1020004) in TMEM106B correlated with increased risk of FTLD-TDP. The expression level of TMEM106B mRNA in the frontal cortex was significantly higher in FTLD-TDP patients, particularly those with GRN mutations, than in healthy controls (HCs)[16], suggesting that genetic variants in TMEM106B are risk factors for FTLD-TDP. Another study reported that the expression of TMEM106B protein in the brains of FTLD-TDP patients with GRN mutations was also higher than that of HCs and other NDs[24]. GRN mutation carriers with homozygotes for the major (risk) allele of rs1990622 showed earlier age at onset and increased disease penetrance than heterozygotes and homozygotes for the minor (protective) allele of rs1990622[53,54]. While another study found no relationship between rs1990622 SNP in TMEM106B and age at the onset of FTLD[55]. The genetic polymorphisms of TMEME106B are associated with lower serum GRN mRNA levels in GRN mutation carriers. There was a negative correlation between TMEME106B mRNA and GRN mRNA levels in the peripheral blood of patients with FTLD[53,54], suggesting that TMEM106B may exert its effects on the phenotypes of FTLD by GRN expression. A neuroimaging study demonstrated that the gray matter volume of several brain regions affected in FTLD (including the frontal and temporal lobes) was significantly smaller in FTLD patients with AA (risk allele of rs1990622 in TMEM106B) than in FTLD patients with AG/GG[18]. The major allele of rs1990621 (another SNP of TMEM106B) correlated with more rapid cognition decline in FTLD patients[20]. However, rs1990621 was identified as a protective variant for FTLD and was associated with increased neuronal proportion in another study[56].
The hexanucleotide repeat GGGGCC in the C9orf72 gene is the most common genetic cause of familial FTLD and ALS[57,58]. Unlike the conditions in FTLD mentioned above, the major allele of rs1990622 in TMEM106B associates with later age of onset and death of FTLD patients with C9orf72 mutation and a later age at death, while TMEM106B rs1990622 SNP shows no influence on age at onset or death of FTLD-TDP patients without GRN or C9orf72 mutation[17]. The genotype of TMEM106B in FTLD-TDP patients appears to protect patients with C9orf72 mutation, suggesting the complexity of TMEM106B regulation in FTLD. In addition, the frequency of the minor allele homozygote of rs3173615 (contributes to the nonsynonymous mutation p.T185S) is obviously reduced in patients with C9orf72 mutation in comparison with HCs, which suggests that those expressing homozygote of the minor allele are less likely to develop FTLD[59,60]. An in vitro study found that Hela cells transfected with the T185 vector showed higher expression of TMEM106B than cells transfected with the S185 vector because the degradation rate of S185-TMEM106B was faster than T185-TMEM106B[61]; these findings suggest a mechanism mediating different risks of FTLD resulting from TMEM106B rs3173615 variants.
TMEM106B in other NDs
Several lines of evidence also suggest a role for TMEM106B in other NDs. Three SNPs of TMEM106B are associated with AD, the most common ND and the leading cause of dementia worldwide[62]. These include rs1595014, rs1990620, and rs1990622 [Table 1]. Although genotype and allele frequencies of rs1990622 do not differ between AD patients and HCs when the status with or without the APOE4 allele is not considered, the frequencies of the major allele homozygote of rs1990622 were significantly higher in AD patients carrying APOE4 allele than HCs. These findings suggest TMEM106B genetic variants might modify AD by interacting with APOE4[63]. A similar study revealed that rs1595014 in TMEM106B is a risk modifier for AD through its mutual effect with APOE4[64]. Another study found that AD patients with the rs1990622 major allele haplotype were more likely to develop TDP-43 pathology than those carrying the minor allele[65], suggesting that the TMEME106B variant influences the pathological phenotype of AD. A bioinformatics analysis demonstrated that the signal pathways associated with immune function and neuroinflammation are upregulated in late-onset AD patients harboring the risk haplotype of rs1990620 in TMEM106B[66]. The TMEM106B expression level was lower in several brain regions and cell types in AD patients than in HCs[25], supporting the implication of TMEM106B in AD pathogenesis.
ALS is a fatal ND affecting motor neurons in the brain and spinal cord; its clinical and pathological features overlap with FTLD-TDP[57]. Similar to the phenomenon observed in FTLD, the major allele of rs1990622 on TMEM106B promotes cognitive decline as measured by the Mini-Mental State Examination and the Montreal Cognitive Assessment in ALS[20,67] and PD[20,68]. However, another study found that patients with ALS harboring the major allele of rs1990622 showed better cognition but worse motor functions than patients homozygous for the minor allele[69]. Among ALS patients, minor allele homozygous carriers developed more severe TDP-43 pathology than major allele homozygotes or major and minor allele heterozygotes with or without adjustment for the C9orf72 mutation[22]. TMEM106B knockdown phenocopied this result in vitro[22], suggesting that variants of TMEM106B affect TDP-43 pathology in ALS by regulating TMEM106B expression levels.
Chronic traumatic encephalopathy (CTE) is an ND most often identified in postmortem autopsies of individuals exposed to repetitive head impacts. The clinical features of CTE are often progressive, leading to dramatic changes in mood, behavior, and cognition, often resulting in debilitating dementia[70]. The characteristic neuropathological findings of CTE include phosphorated-tau accumulations involving superficial cortical layers commonly located at the depths of the cerebral sulci and in perivascular spaces[71]. Among pathologically-identified CTE patients, there were significantly fewer homozygous carriers of the minor allele of rs3173615 in TMEM106B than those without CTE pathology. Moreover, the tau pathology appears to be most severe in homozygous carriers of the major allele, while the pathology was mildest in the homozygous carriers of the minor allele[72]. This finding suggests that TMEM106B variants modify tau pathology in CTE patients. However, another study reported that the genetic variations of rs3173615 in TMEM106B in CTE patients were not distinct from neuropathological negative controls[73]. Nevertheless, among neuropathologically verified CTE patients, the dorsolateral frontal cortex in the minor allele carriers presented slighter phosphorylated tau pathology and neuroinflammation, and higher synaptic protein density than the major allele carriers[73]. In addition, rs1990622 in TMEM106B (the top SNP risk factor identified for FTLD-TDP[16]) increases the risk of developing hippocampal sclerosis of aging (HS-aging)[74,75] and limbic-predominant age-related TDP-43 encephalopathy (LATE)[76,77].
Together, variants of TMEME106B are genetic modifiers of risk for NDs, including AD, ALS, PD, CTE, HS-aging, and LATE. Furthermore, the genotype variations of TMEM106B influence several clinical and pathological phenotypes of these NDs.
TMEM106B in brain aging
Aging is the most critical risk factor for most of NDs[42]. Aging brains without known disorders share characteristics with NDs, including mitochondrial dysfunction, protein homeostasis imbalance, and disturbed intercellular communication[78]. Protein aggregation, thought to be a pathological hallmark for NDs, also occurs in the brains of clinically normal older adults; these aggregates include Aβ plaques, neurofibrillary tangles composed of tau, LBs, and LNs composed of aggregated α-syn, and TDP-43 immunoreactive inclusions[79-81].
Like NDs, the genotype polymorphism and its function were explored in normal aging populations. A study leveraging RNA sequencing data revealed that the temporal cortex of normal older adults with different haplotypes of rs3173615 in TMEM106B had distinct gene expression patterns[82]. Another study found that TMEM106B and GRN variants synergistically influenced the aging brain's transcriptome[83]. Consistent with the results in FTLD, reduced left hemisphere volume was observed in the general population with TMEM106B rs1990622 risk allele[84]. These data suggest the involvement of the TMEM106B genetic variation in brain aging.
IDENTIFICATION AND CRYO-EM STRUCTURE OF TMEM106B FIBRILS
Abnormal cerebral aggregations of pathogenic proteins (Aβ, tau, α-syn, and TDP-43) in NDs were found in the form of amyloid fibrils resistant to sarkosyl[85-88]. Thus, the inclusions composed of pathogenic fibrillar proteins are considered the pathological hallmark of NDs. With the rapid development of equipment and technology of cryo-EM, the atomic structures of brain-extracted Aβ fibrils was elucidated in most NDs[14], tau fibrils in all tauopathies[7-9,11,12,89], α-syn fibrils in common α-synucleinopathies[10,15], and TDP-43 fibrils in ALS were determined[13]. The cryo-EM structure determination of these pathogenic proteins strengthens our understanding of the molecular pathogenesis of NDs and contributes to developing antibodies and small molecules targeting filamentous aggregation to inhibit further aggregates formation or facilitate aggregates degradation. A previously unknown amyloid fibril, formed by the luminal domain of TMEM106B, in the brain of several ND patients and normal older adults was identified by several independent groups[29-32]. Information about all the donors with TMEM106B fibrils from the four groups is summarized in Table 2. Donors with NDs included: AD, including sporadic AD and sporadic early-onset AD; tauopathies, including AGD, aging-related tau astrogliopathy, and CBD; familial frontotemporal dementia and parkinsonism linked to chromosome 17 caused by MAPT mutations (FTDP-17), limbic-predominant neuronal inclusion body 4R tauopathy (LNT)12, primary age-related tauopathy (PART), and PSP; α-synucleinopathies, including sporadic or familial PD, PDD, DLB, and MSA; TDP-43 proteinopathies, including ALS, FTLD-TDP with different subtypes of TDP-43 pathology; pathological aging (PA), and vascular dementia (VaD).
Summaries of information for donors withTMEM106B fibrils
| Case | Disease | Age (yr) | Gender | TMEM106B Polymorphs | FH | T185S SNP | Brain region | Reference |
| 1 | AD | 79 | M | S-I, D-Ia | No | SS | Frontal cortex | Schweighauser et al.[29] |
| 2 | FAD | 67 | F | S-I, D-Ia | Yes | TT | Frontal cortex | Schweighauser et al.[29] |
| 3 | EOAD | 58 | F | S-I, D-Ia | No | TT | Frontal cortex | Schweighauser et al.[29] |
| 4 | PA | 59 | M | S-I, D-Ia | No | TS | Frontal cortex | Schweighauser et al.[29] |
| 5 | CBD | 74 | F | S-I, D-Ia | No | TS | Frontal cortex | Schweighauser et al.[29] |
| 6 | CBD | 79 | F | S-I, D-Ia | No | TS | Frontal cortex | Schweighauser et al.[29] |
| 7 | FTDP-17T | 55 | M | S-I, D-Ia | Yes | TT | Temporal cortex | Schweighauser et al.[29] |
| 8 | AGD | 85 | M | S-III | No | TS | Nucleus accumbens | Schweighauser et al.[29] |
| 9 | AGD | 90 | M | S-I, D-Ia | No | TT | Nucleus accumbens | Schweighauser et al.[29] |
| 10 | LNT | 66 | F | S-I, D-Ia | No | TT | Frontal cortex | Schweighauser et al.[29] |
| 11 | ARTAG | 85 | F | S-III | No | SS | Hippocampus | Schweighauser et al.[29] |
| 12 | PD | 87 | M | S-III | No | SS | Cingulate gyrus | Schweighauser et al.[29] |
| 13 | PDD | 64 | M | S-I, D-Ia | No | TT | Amygdala | Schweighauser et al.[29] |
| 14 | FPD | 67 | NA | S-III | Yes | SS | NA | Schweighauser et al.[29] |
| 15 | DLB | 74 | M | S-III | No | SS | Frontal cortex | Schweighauser et al.[29] |
| 16 | DLB | 73 | M | S-I, D-Ia | No | TS | Frontal cortex | Schweighauser et al.[29] |
| 17 | MSA | 85 | F | S-III | No | SS | Putamen | Schweighauser et al.[29] |
| 18 | MSA | 70 | M | S-I, D-Ia | No | TS | Putamen | Schweighauser et al.[29] |
| 19 | MSA | 68 | F | S-IIa, S-IIb | No | TT | Putamen | Schweighauser et al.[29] |
| 20 | FTLD-TDP-A | 66 | F | S-I, D-Ia | Yes | TS | Frontal cortex | Schweighauser et al.[29] |
| 21 | FTLD-TDP-C | 65 | F | S-III | No | SS | Frontal cortex | Schweighauser et al.[29] |
| 22 | ALS-TDP-B | 63 | F | S-III | No | SS | Motor cortex | Schweighauser et al.[29] |
| 23 | Normal control | 75 | M | S-I, D-Ia | NA | TS | Frontal cortex | Schweighauser et al.[29] |
| 24 | Normal control | 84 | M | S-I, D-Ia | NA | TS | Frontal cortex | Schweighauser et al.[29] |
| 25 | Normal control | 101 | M | S-I, D-Ia | NA | TT | Frontal cortex | Schweighauser et al.[29] |
| 26 | FTLD-TDP-A | 60 | M | S-I, D-Ia | Yes | TT | Frontal cortex | Chang et al.[31] |
| 27 | FTLD-TDP-A | 55 | F | S-I, D-Ia | Unknown | TS | Frontal cortex | Chang et al.[31] |
| 28 | FTLD-TDP-A | 60 | F | D-Ia | Yes | TT | Frontal cortex | Chang et al.[31] |
| 29 | FTLD-TDP-A | 89 | M | S-I, D-Ia | No | TS | Frontal cortex | Chang et al.[31] |
| 30 | FTLD-TDP-A | 48 | F | D-Ia | Yes | TT | Frontal cortex | Chang et al.[31] |
| 31 | FTLD-TDP-B | 62 | M | ? | No | TS | Frontal cortex | Chang et al.[31] |
| 32 | FTLD-TDP-B | 74 | F | S-III | No | SS | Frontal cortex | Chang et al.[31] |
| 33 | FTLD-TDP-C | 69 | M | S-I, D-Ia | No | TT | Frontal cortex | Chang et al.[31] |
| 34 | PSP | 68 | M | S-I | NA | NA | Caudate | Chang et al.[31] |
| 35 | PSP | 75 | M | S-I, D-Ia | No | TS | Frontal cortex | Chang et al.[31] |
| 36 | DLB | 68 | M | S-I, D-Ia | No | NA | Frontal cortex | Chang et al.[31] |
| 37 | FTLD-TDP-A | 86 | M | S-I, D-Ia, D-Ib | No | TT | Medial frontal gyrus | Jiang et al.[30] |
| 38 | FTLD-TDP-B | 76 | F | S-I, D-Ia, D-Ib | No | TS | Medial frontal gyrus | Jiang et al.[30] |
| 39 | FTLD-TDP-C | 65 | M | S-I, D-Ia, D-Ib | No | TS | Medial frontal gyrus | Jiang et al.[30] |
| 40 | FTLD-TDP-D | 64 | F | S-I, D-Ia, D-Ib | Yes | TS | Medial frontal gyrus | Jiang et al.[30] |
| 41 | PDD | 70 | F | S-I, D-Ia | No | NA | Frontal cortex | Fan et al.[32] |
| 42 | Normal control | 71 | M | S-I | NA | NA | Temporal cortex | Fan et al.[32] |
| 43 | Normal control | 101 | M | S-III | NA | NA | Temporal cortex | Fan et al.[32] |
EXTRACTION AND IDENTIFICATION OF TMEM106B FIBRILS
The extraction protocols for TMEM106B fibrils from the four groups were similar, as were the other amyloid fibrils (i.e., Aβ, tau, ad α-syn). Nevertheless, there are some differences, including the time when sarkosyl was added, the concentration of sarkosyl and its incubation time with homogenates, centrifugation speed and time, and the treatment of pronase. The methods used to identify the previously unknown TMEM106B fibrils differed among the studies. Cryo-EM and mass spectrometry were used to identify the protein that forms the previously unsolved amyloid fibrils in Fan et al. and Chang et al.[31,32]. The other two groups adopted model building and specific peptide searching[29,30].
CRYO-EM STRUCTURE OF TMEM106B POLYMORPHS
Six polymorphs with three folds (called folds I, IIa, IIb, and III) of TMEM106B fibrils were found in human brains [Figure 1A and B, Table 2]. Four polymorphs consist of single protofilament (S-I, S-IIa, S-IIb, and S-III), and the remaining two polymorphs comprise double protofilaments of fold I (D-Ia and D-Ib). The fibril core of all polymorphs is composed of residues 120-254 of TMEM106B, forming 17-19 β-strands and folds into a five-layer structure. The three folds’ structure is divided into the N-terminal region (S120-T166), the middle region (A167-M210), and the C-terminal region (Y211-G254), which form the first two layers, the fifth layer, and the central two layers of the well-ordered fibril core, respectively. There are two subtypes of fold II (IIa, IIb) because of the difference in A167-I187.
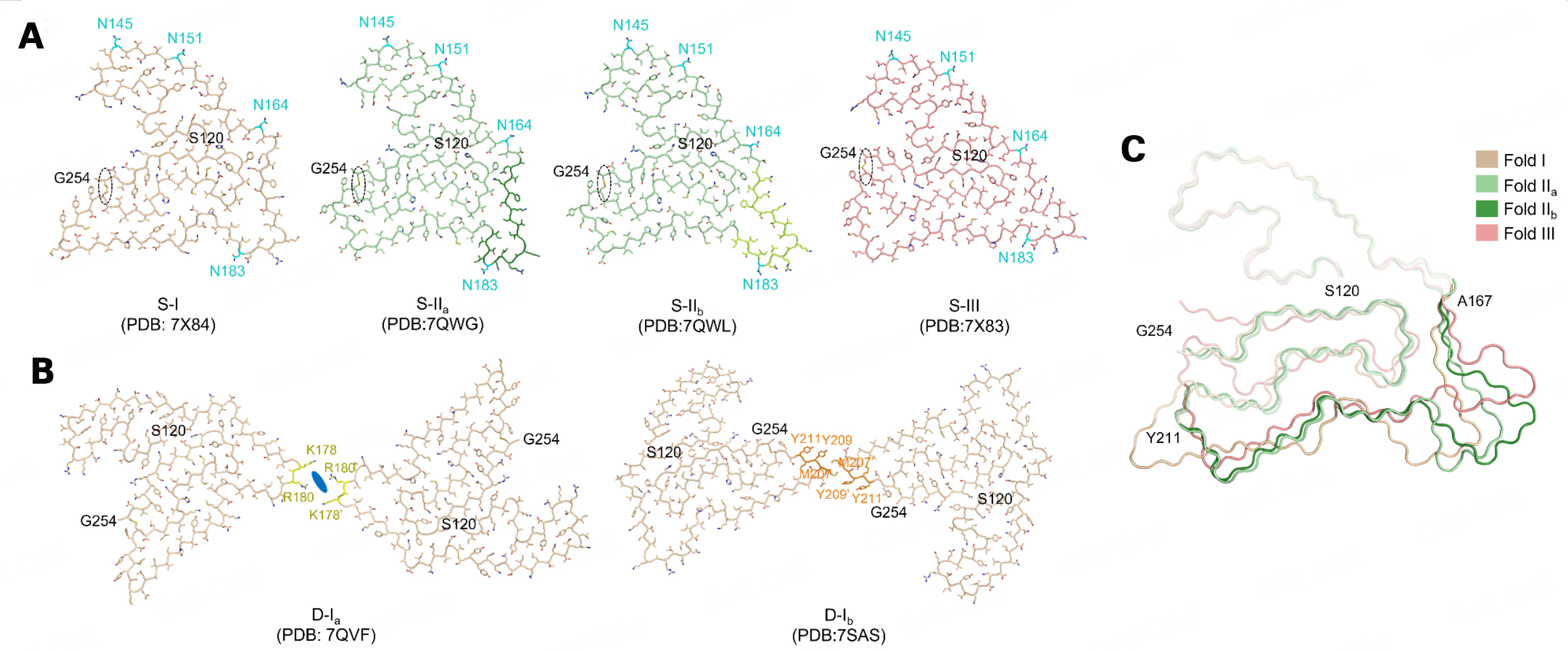
Figure 1. Polymorphs of ex vivo TMEM106B fibrils. A: Singlet of ex vivo TMEM106B polymorphs. The residues with glycosylation, N145, N151, N164, and N183, are highlighted in cyan. The dashed oval indicates the disulfide bond between C214 and C253; B: Doublet of ex vivo TMEM106B polymorphs. The residues comprising the interface of D-Ia and D-Ib are highlighted in yellow and orange, respectively. The blue oral indicates the unknown density comprising the D-Ia interface; C: Overlay of Folds I, IIa, IIb, and III. The N-terminal region (S120-T166), the middle region (A167-M210), and the C-terminal region (Y211-G254) of the three folds are indicated by different transparency. Side chains in (A) and (B) are shown as sticks.
All polymorphs share two structural characteristics. One is the glycosylation at N145, N151, N164, and N183; the other is the disulfide bond formed between C214 and C253. Four singlets share the same N-terminal region and present distinct structures in the middle region and slight structural differences in the C-terminal region [Figure 1C]. Both doublets comprise two protofilaments of fold I but with different interfaces [Figure 1B]. The interface of D-Ia consists of two positively-charged amino acids (K178 and R180) of the two protofilaments and an unknown additional density (possibly an anion), whereas that of D-Ib composes of a hydrophobic interaction between M207 and Y209.
The TMEM106B fibril formation process is unknown. An in vitro study found that TMEM106B could be cleaved into the luminal domain and an N-terminal fragment by unknown lysosomal proteases at uncertain amino sites and is further cleaved by SPPL2a, resulting in the generation of the intracellular cytosolic domain[90]. In the native state, CTD of TMEM106B is predicted to form seven β-sheets and is prone to aggregate[91]. In the fibrillar state, TMEM106B consists mainly of the CTD (120-254aa) but with β-sheets rearranged. The N-terminal Ser120 is deeply buried inside the TMEM106B fibril core, precluding additional undetermined residues. Therefore, the identification of TMEM106B fibrils suggests two implications. One is that only CTD is enriched in TMEM106B aggregates, and the other is that before forming amyloid aggregates, the luminal domain is cleaved off at Arg119.
POLYMORPHISM COMPARISONS BETWEEN TMEM106B FIBRILS AND OTHER AMYLOID FIBRILS
Cryo-EM and solid-state nuclear magnetic resonance studies revealed that different proteins or the same protein could form amyloid fibrils with several structural polymorphs in different diseases or under different in vitro conditions [Figure 2]. For example, ex vivo tau fibrils from various tauopathies show distinct folds and represent different polymorphs, on which basis the classification of tauopathies based on the biological conformation of tau fibrils was established[7-9,11,12,89]. Similarly, α-syn forms many fibril polymorphs in different α-synucleinopathies and under different in vitro conditions[10,15,92-94]. In contrast, although TMEM106B can form polymorphic fibrils, different TMEM106B polymorphs share a similar curling stone-like fold. The limited folding of TMEM106B may be related to its extensive glycosylation at Asn residues, the preformed disulfide bond, and other potential PTMs. Moreover, no relationship between the fold of TMEM106B and NDs was observed. For example, fold I could be found in AD, CBD, PDD, DLB, MSA, PSP, PA, AGD, LNT, FTDP-17T, FTLD-TDP, and normal older adults. The same ND could possess different TMEM106B folds; for example, all three folds were observed in MSA.
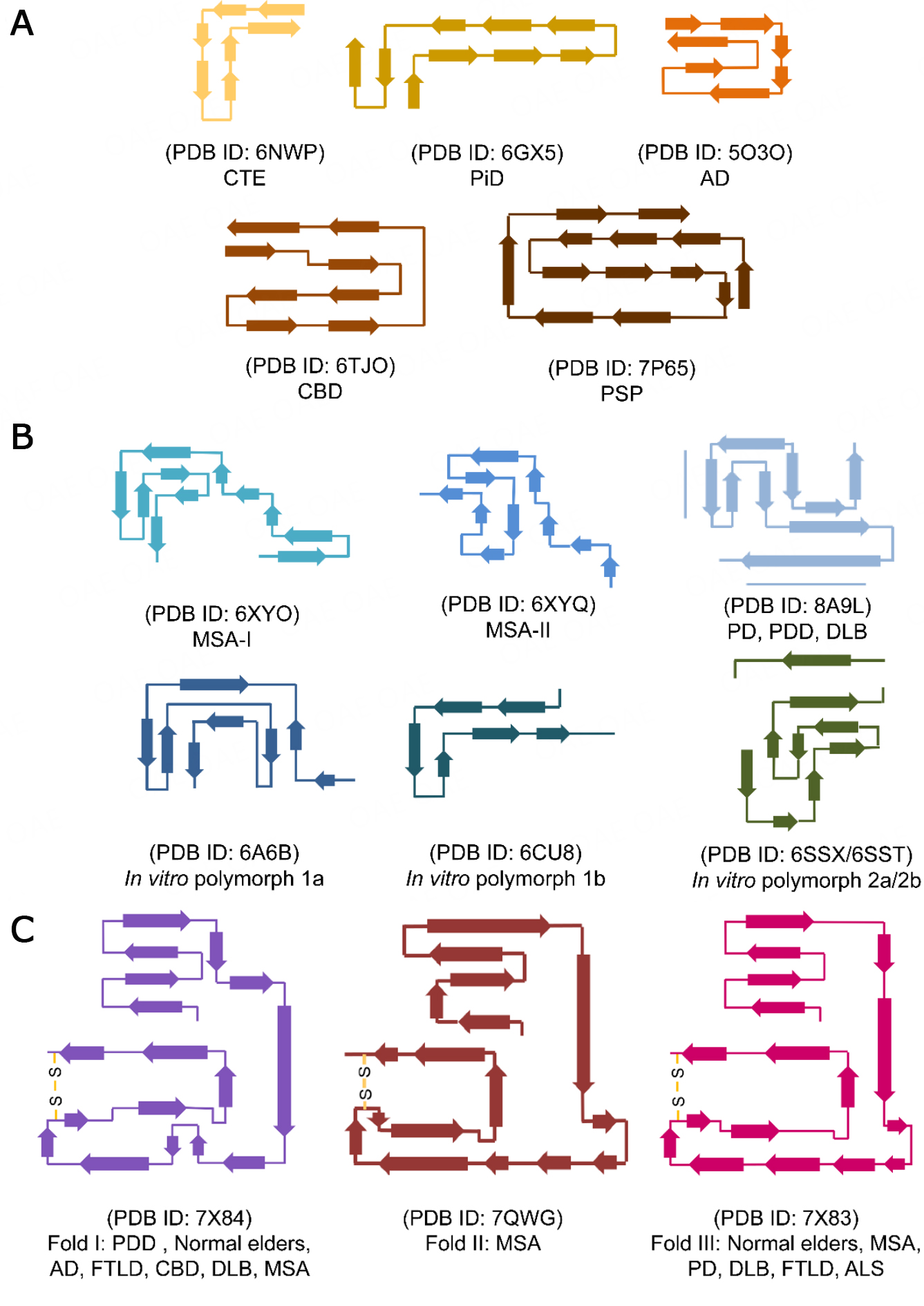
Figure 2. Comparison of the structural polymorphism of amyloid fibrils formed by tau, α-syn, and TMEM106B. The topology diagrams show different polymorphic structures of brain-extracted tau fibrils (A), brain-extracted and in vitro assembled α-syn fibrils (B), and brain-extracted TMEM106B fibrils (C). AD: Alzheimer’s disease; ALS: amyotrophic lateral sclerosis; CBD: corticobasal degeneration; CTE: chronic traumatic encephalopathy; DLB: dementia with Lewy bodies; FTLD: frontotemporal lobar degeneration; MSA: multiple system atrophy; PD: Parkinson's disease; PDD: Parkinson's disease with dementia; PiD: Pick's disease; PSP: progressive supranuclear palsy.
TMEM106B POLYMORPHISM IS ASSOCIATED WITH TMEM106B VARIATION
The minor allele of rs3173615 contributing to the nonsynonymous mutation p.T185S in TMEM106B was identified as a protective factor for FTLD[60,61]. The genetic phenotypes of p.T185S were recorded in 38 of 43 donors with TMEM106B fibrils. The TT homozygotes, TS heterozygotes, and SS homozygotes accounted for 34%, 42%, and 24% percent, respectively [Figure 3A]. Interestingly, similar to the genetic variation of TMEM106B, the distribution of TMEM106B fibril folds differs, with fold I being the most common, followed by folds III and II [Figure 3B]. Moreover, though independent from conditions of NDs, the folds of TMEM106B fibrils are associated with TMEM106B p.T185S variation. Specifically, fold I predominantly exists in individuals with TT and TS, while fold III predominates in subjects with SS [Figure 3C]. Thus, TMEM106B p.T185S variation is critical in forming different TMEM106B fibril folds. There is clinicopathological heterogeneity across patients with NDs carrying different alleles of TMEM106B p.T185S and the degradation variation of T185-TMEM106B protein and S185-TMEM106B protein[60,61]; future research should address whether the formation of the three folds differs and whether the three folds exert different influences on ND pathogenesis and the aging process.
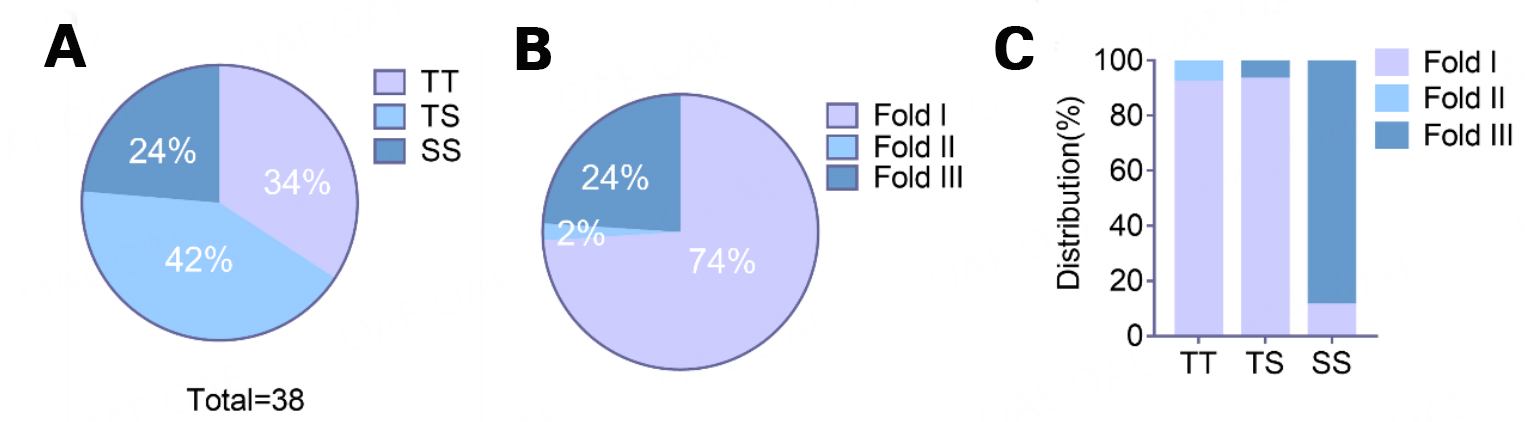
Figure 3. The distribution of TMEM106B p.T185S variation and TMEM106B fibril fold. A: TMEM106B p.T185S variation distribution in 38 donors with TMEM106B fibrils. B: TMEM106B fibril folds distribution in patients with NDs and normal older adults. C: TMEM106B fibril folds distribution in donors with different TMEM106B p.T185S variations.
IMPLICATIONS AND FUTURE DIRECTIONS
Unsurprisingly, TMEM106B aggregates into amyloid fibrils in several NDs and normal older subjects because its genetic variation is associated with several clinicopathological phenotypes of NDs and normal older brains. However, different, even contradictory, conclusions about the TMEM106B fibrils were made by different groups. Because TMEM106B fibrils were found in older individuals with or without NDs but not in younger subjects, Schweighauser et al. concluded that TMEM106B fibrils are age-dependent and unrelated to diseases[29]. In contrast, Jiang et al. insisted on the pathological role of TMEM106B fibrils in FTLD-TDP because they found TMEM106B fibrils only in patients with FTLD-TDP and not in normal older adults and patients with VaD or PART[30]. To investigate the implication of TMEM106B fibrils, Fan et al. summarized information from donors from four studies and found that the age of donors with NDs was significantly younger than that of normal older adults, suggesting a correlation of TMEM106B fibril formation with NDs[32]. Nevertheless, based on the scant data about TMEM106B fibrils in the pathogenesis of NDs, the links between TMEM106B aggregates and other pathogenic proteins of NDs remain unknown.
Because the genotype of TMEM106B is related to clinicopathologies of NDs and contributes to the polymorphism of ex vivo TMEM106B fibrils, it is unlikely that TMEM106B fibrils are simply by-products of aging. Though TMEM106B fibrils also form in normal older adults, we could not exclude its potential pathogenic effects because aging itself is a primary risk factor for NDs[42]; pathogenic fibrillar proteins such as α-syn, tau, and TDP-43 are also present in aged individuals without NDs[79,80,95,96]. Indeed, the finding of TMEM106B fibrils would evoke enthusiasm in functional studies of TMEM106B in aging and diseased conditions.
The function of TMEM106B fibrils could be explored from the following aspects in the future. First, it is essential to determine whether TMEM106B fibrils influence the positron emission tomography imaging for amyloid aggregates such as Aβ and tau in NDs. Second, given that TMEM106B fibrils are present in the brains of NDs and normal older adults, it is vital to determine whether TMEM106B fibrils are pathological aggregates or simply by-products of normal aging. Therefore, future studies need to determine whether TMEM106B fibrils are neurotoxic and cause neurodegeneration, as other pathological amyloid fibrils do. Third, because TMEM106B amyloid fibrils were recently identified in the human brain, the distribution pattern of TMEM106B aggregates in the brain of normal older adults and NDs should be established, as Braak staging of α-syn pathology in PD and tau pathology in AD did[97,98]. In addition, it is essential to determine whether TMEM106B aggregates are co-pathologies of other pathological amyloid aggregates in the brain of NDs. Finally, it is critical to measure TMEM106B levels in biological samples such as cerebral spinal fluid and serum from patients with NDs and age- and sex-matched HCs; doing so will determine whether TMEM106B could be a potential biomarker for ND diagnosis.
In addition to functional exploration, several other questions about TMEM106B fibrils need to be investigated. First, in vitro replications of the TMEM106B fibril experiments must be performed to elucidate the molecular mechanisms and conditions for the fibril formation. Second, the relationship or interaction between TMEM106B aggregation and other pathogenic proteins of NDs needs to be studied. Finally, current antibodies were designed for native TMEM106B protein, and there is an urgent need for antibodies specifically targeting TMEM106B inclusions.
CONCLUSION
TMEM106B is a type II membrane protein that participates crucially in lysosome morphology, intracellular localization, trafficking, and acidification. It forms amyloid fibrils in the brains of patients with many NDs and neurologically normal older adults. Because genotype variation of TMEM106B is associated with the clinicopathological phenotypes of multiple NDs and contributes to the polymorphism of TMEM106B fibrils, it is plausible to speculate that TMEM106B fibrils are possible pathogens rather than just by-products produced during the development and progression of NDs and aging. It is also possible that the polymorphisms of TMEM106B fibrils resulting from the genetic variation of TMEM106B play critical roles in the clinicopathological heterogeneity of NDs. The investigations of the functions and roles of TMEM106B fibrils in NDs and aging are urgently needed.
DECLARATIONS
AcknowledgmentsThe authors gratefully acknowledge Zhao QY for assisting with preparing Figure 2 and Liu Q for editing the manuscript.
Author’s contributionsWrote the manuscript: Fan Y, Zhao W
All authors contributed to the manuscript discussion and editing: Fan Y, Zhao W, Ni Y, Liu Y, Tang Y, Sun Y, Liu F, Yu W, Wu J, Wang J
Supervised the manuscript writing: Wang J
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by The National Natural Science Foundation of China (Grant No. 82171421, 91949118, and 81771372), the National Health Commission of China (Pro20211231084249000238), the Science and Technology Commission of Shanghai Municipality (Grant No. 21S31902200), the Shanghai Municipal Science and Technology Major Project (Grant No. 2018SHZDZX01), the ZJ Lab, and the Shanghai Center for Brain Science and Brain-Inspired Technology.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2023.
REFERENCES
1. Goedert M. NEURODEGENERATION. Alzheimer's and Parkinson's diseases: The prion concept in relation to assembled Aβ, tau, and α-synuclein. Science 2015;349:1255555.
2. Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006;314:130-3.
3. Peng C, Gathagan RJ, Lee VM. Distinct α-Synuclein strains and implications for heterogeneity among α-Synucleinopathies. Neurobiol Dis 2018;109:209-18.
4. Peng C, Gathagan RJ, Covell DJ, et al. Cellular milieu imparts distinct pathological α-synuclein strains in α-synucleinopathies. Nature 2018;557:558-63.
5. Lau A, So RWL, Lau HHC, et al. α-Synuclein strains target distinct brain regions and cell types. Nat Neurosci 2020;23:21-31.
6. Nonaka T, Masuda-Suzukake M, Arai T, et al. Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep 2013;4:124-34.
7. Fitzpatrick AWP, Falcon B, He S, et al. Cryo-EM structures of tau filaments from Alzheimer's disease. Nature 2017;547:185-90.
8. Falcon B, Zhang W, Murzin AG, et al. Structures of filaments from Pick's disease reveal a novel tau protein fold. Nature 2018;561:137-40.
9. Falcon B, Zivanov J, Zhang W, et al. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature 2019;568:420-3.
10. Schweighauser M, Shi Y, Tarutani A, et al. Structures of α-synuclein filaments from multiple system atrophy. Nature 2020;585:464-9.
11. Zhang W, Tarutani A, Newell KL, et al. Novel tau filament fold in corticobasal degeneration. Nature 2020;580:283-7.
12. Shi Y, Zhang W, Yang Y, et al. Structure-based classification of tauopathies. Nature 2021;598:359-63.
13. Arseni D, Hasegawa M, Murzin AG, et al. Structure of pathological TDP-43 filaments from ALS with FTLD. Nature 2022;601:139-43.
14. Yang Y, Arseni D, Zhang W, et al. Cryo-EM structures of amyloid-β 42 filaments from human brains. Science 2022;375:167-72.
15. Yang Y, Shi Y, Schweighauser M, et al. Structures of α-synuclein filaments from human brains with Lewy pathology. Nature 2022;610:791-5.
16. Van Deerlin VM, Sleiman PM, Martinez-Lage M, et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet 2010;42:234-9.
17. Gallagher MD, Suh E, Grossman M, et al.
18. Harding SR, Bocchetta M, Gordon E, et al. The TMEM106B risk allele is associated with lower cortical volumes in a clinically diagnosed frontotemporal dementia cohort. J Neurol Neurosurg Psychiatry 2017;88:997-8.
19. Pottier C, Zhou X, Perkerson RB 3rd, et al. Potential genetic modifiers of disease risk and age at onset in patients with frontotemporal lobar degeneration and GRN mutations: a genome-wide association study. Lancet Neurol 2018;17:548-58.
20. Tropea TF, Mak J, Guo MH, et al.
21. Hu Y, Sun JY, Zhang Y, et al. rs1990622 variant associates with Alzheimer's disease and regulates
22. Mao F, Robinson JL, Unger T, et al. TMEM106B modifies TDP-43 pathology in human ALS brain and cell-based models of TDP-43 proteinopathy. Acta Neuropathol 2021;142:629-42.
23. Cykowski MD, Arumanayagam AS, Powell SZ, et al. Patterns of amygdala region pathology in LATE-NC: subtypes that differ with regard to TDP-43 histopathology, genetic risk factors, and comorbid pathologies. Acta Neuropathol 2022;143:531-45.
24. Busch JI, Martinez-Lage M, Ashbridge E, et al. Expression of TMEM106B, the frontotemporal lobar degeneration-associated protein, in normal and diseased human brain. Acta Neuropathol Commun 2013;1:36.
25. Satoh J, Kino Y, Kawana N, et al. TMEM106B expression is reduced in Alzheimer's disease brains. Alzheimers Res Ther 2014;6:17.
26. Brady OA, Zheng Y, Murphy K, Huang M, Hu F. The frontotemporal lobar degeneration risk factor, TMEM106B, regulates lysosomal morphology and function. Hum Mol Genet 2013;22:685-95.
27. Schwenk BM, Lang CM, Hogl S, et al. The FTLD risk factor TMEM106B and MAP6 control dendritic trafficking of lysosomes. EMBO J 2014;33:450-67.
28. Klein ZA, Takahashi H, Ma M, et al. Loss of TMEM106B ameliorates lysosomal and frontotemporal dementia-related phenotypes in progranulin-deficient mice. Neuron 2017;95:281-296.e6.
29. Schweighauser M, Arseni D, Bacioglu M, et al. Age-dependent formation of TMEM106B amyloid filaments in human brains. Nature 2022;605:310-4.
30. Jiang YX, Cao Q, Sawaya MR, et al. Amyloid fibrils in FTLD-TDP are composed of TMEM106B and not TDP-43. Nature 2022;605:304-9.
31. Chang A, Xiang X, Wang J, et al. Homotypic fibrillization of TMEM106B across diverse neurodegenerative diseases. Cell 2022;185:1346-1355.e15.
32. Fan Y, Zhao Q, Xia W, et al. Generic amyloid fibrillation of TMEM106B in patient with Parkinson's disease dementia and normal elders. Cell Res 2022;32:585-8.
33. Lang CM, Fellerer K, Schwenk BM, et al. Membrane orientation and subcellular localization of transmembrane protein 106B (TMEM106B), a major risk factor for frontotemporal lobar degeneration. J Biol Chem 2012;287:19355-65.
34. Chen-Plotkin AS, Unger TL, Gallagher MD, et al.
35. Feng T, Sheng RR, Solé-Domènech S, et al. A role of the frontotemporal lobar degeneration risk factor TMEM106B in myelination. Brain 2020;143:2255-71.
36. Simons C, Dyment D, Bent SJ, et al. Care4Rare Consortium. A recurrent
37. Kang J, Lim L, Song J. TMEM106B, a risk factor for FTLD and aging, has an intrinsically disordered cytoplasmic domain. PLoS One 2018;13:e0205856.
38. Stagi M, Klein ZA, Gould TJ, Bewersdorf J, Strittmatter SM. Lysosome size, motility and stress response regulated by fronto-temporal dementia modifier TMEM106B. Mol Cell Neurosci 2014;61:226-40.
39. Busch JI, Unger TL, Jain N, Tyler Skrinak R, Charan RA, Chen-Plotkin AS. Increased expression of the frontotemporal dementia risk factor TMEM106B causes C9orf72-dependent alterations in lysosomes. Hum Mol Genet 2016;25:2681-97.
40. Solé-Domènech S, Cruz DL, Capetillo-Zarate E, Maxfield FR. The endocytic pathway in microglia during health, aging and Alzheimer's disease. Ageing Res Rev 2016;32:89-103.
41. Lie PPY, Nixon RA. Lysosome trafficking and signaling in health and neurodegenerative diseases. Neurobiol Dis 2019;122:94-105.
42. Hou Y, Dan X, Babbar M, et al. Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol 2019;15:565-81.
43. Mohandas E, Rajmohan V. Frontotemporal dementia: an updated overview. Indian J Psychiatry 2009; 51 Suppl 1(Suppl1):S65-9.
44. Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 1998;51:1546-54.
45. Goldman JS, Farmer JM, Van Deerlin VM, Wilhelmsen KC, Miller BL, Grossman M. Frontotemporal dementia: genetics and genetic counseling dilemmas. Neurologist 2004;10:227-34.
46. Mackenzie IR, Neumann M, Baborie A, et al. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol 2011;122:111-3.
47. Lee EB, Porta S, Michael Baer G, et al. Expansion of the classification of FTLD-TDP: distinct pathology associated with rapidly progressive frontotemporal degeneration. Acta Neuropathol 2017;134:65-78.
48. Rohrer JD, Guerreiro R, Vandrovcova J, et al. The heritability and genetics of frontotemporal lobar degeneration. Neurology 2009;73:1451-6.
49. Huber N, Korhonen S, Hoffmann D, et al. Deficient neurotransmitter systems and synaptic function in frontotemporal lobar degeneration-Insights into disease mechanisms and current therapeutic approaches. Mol Psychiatry 2022;27:1300-9.
50. Majounie E, Renton AE, Mok K, et al. Chromosome 9-ALS/FTD Consortium. Frequency of the
51. DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of
52. Renton AE, Majounie E, Waite A, et al. ITALSGEN Consortium. A hexanucleotide repeat expansion in
53. Cruchaga C, Graff C, Chiang HH, et al. Association of TMEM106B gene polymorphism with age at onset in granulin mutation carriers and plasma granulin protein levels. Arch Neurol 2011;68:581-6.
54. Finch N, Carrasquillo MM, Baker M, et al.
55. van der Zee J, Van Langenhove T, Kleinberger G, et al.
56. Li Z, Farias FHG, Dube U, et al. The
59. van Blitterswijk M, Mullen B, Nicholson AM, et al.
60. Deming Y, Cruchaga C.
61. Nicholson AM, Finch NA, Wojtas A, et al. TMEM106B p.T185S regulates TMEM106B protein levels: implications for frontotemporal dementia. J Neurochem 2013;126:781-91.
63. Lu RC, Wang H, Tan MS, Yu JT, Tan L. TMEM106B and APOE polymorphisms interact to confer risk for late-onset Alzheimer's disease in Han Chinese. J Neural Transm (Vienna) 2014;121:283-7.
64. Jun G, Ibrahim-Verbaas CA, Vronskaya M, et al. IGAP Consortium. A novel Alzheimer disease locus located near the gene encoding tau protein. Mol Psychiatry 2016;21:108-17.
65. Rutherford NJ, Carrasquillo MM, Li M, et al.
66. Milind N, Preuss C, Haber A, et al. Transcriptomic stratification of late-onset Alzheimer's cases reveals novel genetic modifiers of disease pathology. PLoS Genet 2020;16:e1008775.
67. Vass R, Ashbridge E, Geser F, et al. Risk genotypes at
68. Lemprière S. Frontotemporal dementia risk variant accelerates cognitive decline in Parkinson disease. Nat Rev Neurol 2019;15:307.
69. Manini A, Ratti A, Brusati A, et al.
70. Montenigro PH, Corp DT, Stein TD, Cantu RC, Stern RA. Chronic traumatic encephalopathy: historical origins and current perspective. Annu Rev Clin Psychol 2015;11:309-30.
71. McKee AC, Stern RA, Nowinski CJ, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain 2013;136:43-64.
72. Bieniek KF, Ross OA, Cormier KA, et al. Chronic traumatic encephalopathy pathology in a neurodegenerative disorders brain bank. Acta Neuropathol 2015;130:877-89.
73. Cherry JD, Mez J, Crary JF, et al. Variation in
74. Nelson PT, Wang WX, Partch AB, et al. Reassessment of risk genotypes (
75. Dickson DW, Rademakers R, Nicholson AM, Schneider JA, Yu L, Bennett DA. The
76. Hokkanen SRK, Kero M, Kaivola K, et al. EClipSE Collaboration. Putative risk alleles for LATE-NC with hippocampal sclerosis in population-representative autopsy cohorts. Brain Pathol 2020;30:364-72.
77. Nelson PT, Dickson DW, Trojanowski JQ, et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain 2019;142:1503-27.
78. Culig L, Chu X, Bohr VA. Neurogenesis in aging and age-related neurodegenerative diseases. Ageing Res Rev 2022;78:101636.
79. Crary JF, Trojanowski JQ, Schneider JA, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 2014;128:755-66.
80. Jellinger KA. Lewy body-related alpha-synucleinopathy in the aged human brain. J Neural Transm (Vienna) 2004;111:1219-35.
81. Marks SM, Lockhart SN, Baker SL, Jagust WJ. Tau and β-amyloid are associated with medial temporal lobe structure, function, and memory encoding in normal aging. J Neurosci 2017;37:3192-201.
82. Ren Y, van Blitterswijk M, Allen M, et al.
83. Yang HS, White CC, Klein HU, et al. Genetics of gene expression in the aging human brain reveal TDP-43 proteinopathy pathophysiology. Neuron 2020;107:496-508.e6.
84. Adams HH, Verhaaren BF, Vrooman HA, et al.
85. Goedert M, Spillantini MG, Cairns NJ, Crowther RA. Tau proteins of Alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron 1992;8:159-68.
86. Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. Alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with lewy bodies. Proc Natl Acad Sci USA 1998;95:6469-73.
87. Spillantini MG, Crowther RA, Jakes R, Cairns NJ, Lantos PL, Goedert M. Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson's disease and dementia with Lewy bodies. Neurosci Lett 1998;251:205-8.
88. Crowther RA, Daniel SE, Goedert M. Characterisation of isolated alpha-synuclein filaments from substantia nigra of Parkinson's disease brain. Neurosci Lett 2000;292:128-30.
89. Falcon B, Zhang W, Schweighauser M, et al. Tau filaments from multiple cases of sporadic and inherited Alzheimer's disease adopt a common fold. Acta Neuropathol 2018;136:699-708.
90. Brady OA, Zhou X, Hu F. Regulated intramembrane proteolysis of the frontotemporal lobar degeneration risk factor, TMEM106B, by signal peptide peptidase-like 2a (SPPL2a). J Biol Chem 2014;289:19670-80.
91. Tunyasuvunakool K, Adler J, Wu Z, et al. Highly accurate protein structure prediction for the human proteome. Nature 2021;596:590-6.
92. Li Y, Zhao C, Luo F, et al. Amyloid fibril structure of α-synuclein determined by cryo-electron microscopy. Cell Res 2018;28:897-903.
93. Li B, Ge P, Murray KA, et al. Cryo-EM of full-length α-synuclein reveals fibril polymorphs with a common structural kernel. Nat Commun 2018;9:3609.
94. Guerrero-Ferreira R, Taylor NM, Arteni AA, et al. Two new polymorphic structures of human full-length alpha-synuclein fibrils solved by cryo-electron microscopy. Elife 2019:8.
95. Jellinger KA. Lewy body/alpha-synucleinopathy in schizophrenia and depression: a preliminary neuropathological study. Acta Neuropathol 2009;117:423-7.
96. Yu L, De Jager PL, Yang J, Trojanowski JQ, Bennett DA, Schneider JA. The
97. Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 2003;24:197-211.
98. Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991;82:239-59.
99. Premi E, Formenti A, Gazzina S, et al. Effect of
100. Premi E, Grassi M, van Swieten J, et al. Genetic FTD Initiative (GENFI). Cognitive reserve and
101. Llibre-Guerra JJ, Lee SE, Suemoto CK, et al. A novel temporal-predominant neuro-astroglial tauopathy associated with
102. Murray ME, Cannon A, Graff-Radford NR, et al. Differential clinicopathologic and genetic features of late-onset amnestic dementias. Acta Neuropathol 2014;128:411-21.
103. Hong S, Dobricic V, Ohlei O, et al. Alzheimer's Disease Neuroimaging Initiative (ADNI). TMEM106B and CPOX are genetic determinants of cerebrospinal fluid Alzheimer's disease biomarker levels. Alzheimers Dement 2021;17:1628-40.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Fan Y, Zhao W, Ni Y, Liu Y, Tang Y, Sun Y, Liu F, Yu W, Wu J, Wang J. Newly identified transmembrane protein 106B amyloid fibrils in the human brain: pathogens or by-products?. Ageing Neur Dis 2023;3:4. http://dx.doi.org/10.20517/and.2022.30
AMA Style
Fan Y, Zhao W, Ni Y, Liu Y, Tang Y, Sun Y, Liu F, Yu W, Wu J, Wang J. Newly identified transmembrane protein 106B amyloid fibrils in the human brain: pathogens or by-products?. Ageing and Neurodegenerative Diseases. 2023; 3(1): 4. http://dx.doi.org/10.20517/and.2022.30
Chicago/Turabian Style
Fan, Yun, Wanbing Zhao, You Ni, Yiqi Liu, Yilin Tang, Yimin Sun, Fengtao Liu, Wenbo Yu, Jianjun Wu, Jian Wang. 2023. "Newly identified transmembrane protein 106B amyloid fibrils in the human brain: pathogens or by-products?" Ageing and Neurodegenerative Diseases. 3, no.1: 4. http://dx.doi.org/10.20517/and.2022.30
ACS Style
Fan, Y.; Zhao W.; Ni Y.; Liu Y.; Tang Y.; Sun Y.; Liu F.; Yu W.; Wu J.; Wang J. Newly identified transmembrane protein 106B amyloid fibrils in the human brain: pathogens or by-products?. Ageing. Neur. Dis. 2023, 3, 4. http://dx.doi.org/10.20517/and.2022.30
About This Article
Copyright

Data & Comments
Data
0

 Cite This Article 5 clicks
Cite This Article 5 clicks

 Like This Article 18
likes
Like This Article 18
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.