
Aging and Parkinson's disease: a complex interplay of vulnerable neurons, the immune system and the blood-brain barrier
 0
0 , ...
, ... Abstract
Aging is the biggest risk factor for Parkinson’s disease (PD) and a particular vulnerability of dopaminergic neurons in the substantia nigra to aging-associated effects has been firmly established. More recent work has revealed an important role of non-neuronal systems such as the blood-brain barrier (BBB) or the immune system in the pathogenesis of PD. Effects of aging on the immune system include a chronic inflammatory state termed inflammaging and immunosenescence. Both processes are connected to a higher pro-inflammatory potency and negatively affect the maintenance of self-tolerance. The BBB gets increasingly dysfunctional with advancing age and its endothelial cells display a more pro-inflammatory phenotype while the transport of important plasma proteins to the brain is impaired. The immune system and the BBB are heavily interdependent and are both essential for the homeostasis of especially vulnerable dopaminergic neurons. The degeneration of dopaminergic neurons can, in turn, influence the BBB or the immune system, potentially creating a vicious cycle. In this review, we aim to develop a multisystem perspective on aging and PD by incorporating the aging immune system and aging BBB into the pathophysiological processes. Given the current evidence, it seems likely that a combination of multimodal effects of aging on the levels of SN pars compacta (SNc) dopaminergic neurons, the immune system, and the BBB increase the risk of developing PD.
Keywords
INTRODUCTION
Aging has been shown to be the greatest risk factor for developing idiopathic Parkinson’s disease (PD). The connection between age and PD is best highlighted by the steady increases in disease prevalence among higher age groups, rising from 0.3% for people aged 55 to 64 years to 4.3% for those between 85 and 94 years[1]. This clear relationship between age and prevalence, which was initially established in the Rotterdam Study in the Netherlands, has since been confirmed in multiple cohorts in different countries[2]. Against the background of a rising life expectancy in the general population[3], understanding the implications of this relationship becomes more important and could help identify new molecular targets and develop new therapeutic interventions. In this review, we will elucidate common mechanisms and points of connection between aging and neurodegeneration in PD while trying to establish a framework for further investigation.
In light of recent work in the field, we will focus on the role of dopaminergic neurons, the immune system, and the blood-brain barrier (BBB), which are all implicated in the pathogenesis of PD and undergo profound changes during the process of aging.
Figure 1 gives an overview and a summary of the concepts discussed in this review.
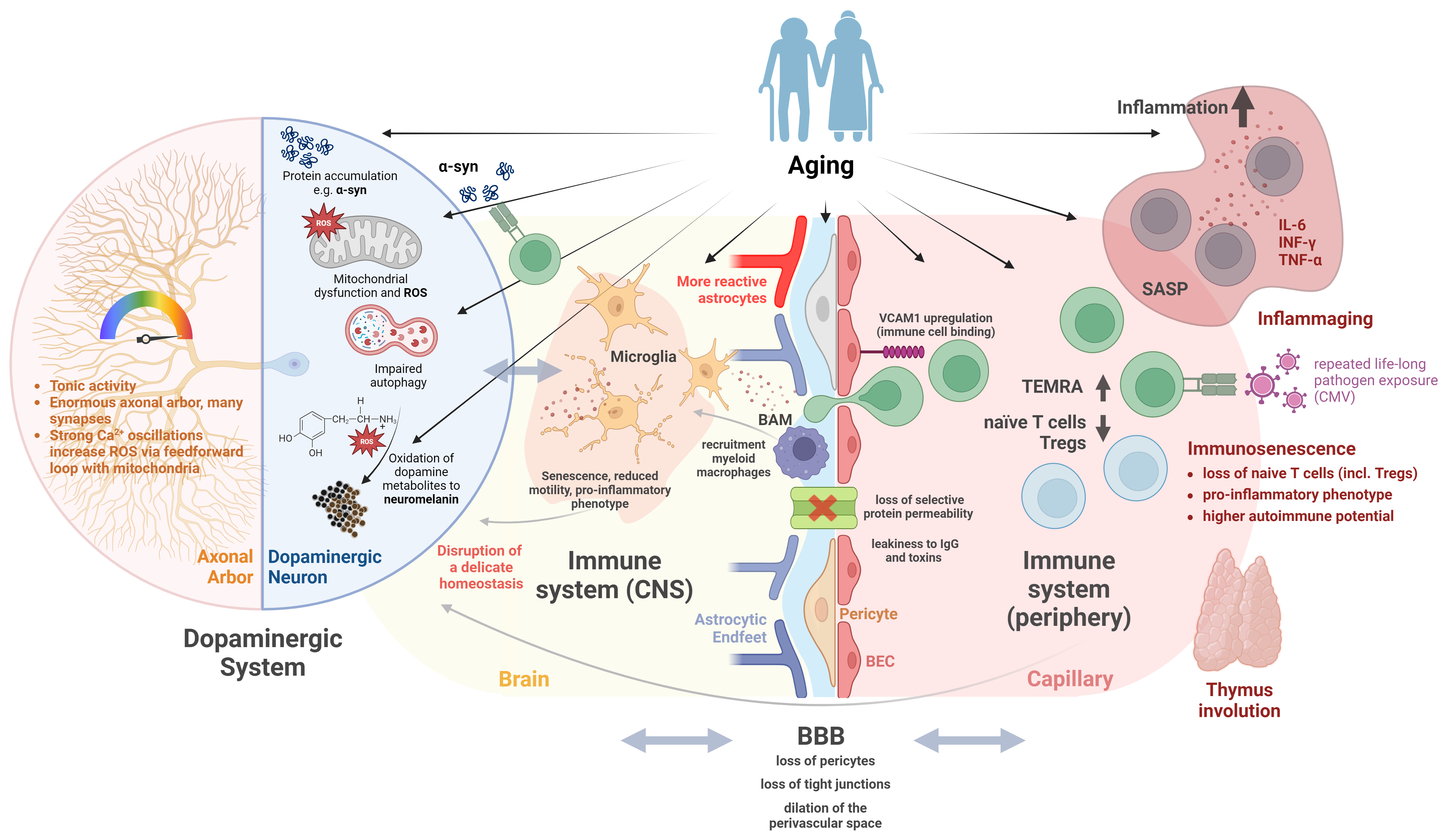
Figure 1. Aging in combination with the particular vulnerability of the dopaminergic system contributes to the pathogenesis of PD. Dopaminergic neurons in the substantia nigra are particularly vulnerable to aging-related alterations in cell metabolism. The sheer size of their axonal arbor in comparison to the soma, and their tonic activity with high cytosolic calcium fluctuation demand high levels of energy for maintenance. Aging-associated mitochondrial insufficiency, impaired protein turnover, and aggregation of proteins lead to additional cell stress via ROS and the accumulation of toxic dopamine metabolites pushing dopamine neurons over the brink of their fragile homeostatic equilibrium. The degeneration of dopaminergic neurons in turn will result in the activation of residing immune cells (microglia), which themselves, with age, tend to present a more pro-inflammatory phenotype, which can perpetuate a hostile environment for neurons. On the other hand, aging itself leads to increased inflammatory microglial phenotype with reduced phagocytic capacity, which attracts peripheral lymphocytes, thereby maintaining an inflammatory environment. With age, parenchymal microglia are also increasingly replaced by peripherally recruited myeloid macrophages, which also tend to have a more pro-inflammatory phenotype. The same is true for the second important tissue-resident macrophage population of the brain, BAMs in the perivascular and meningeal space. Vessel-associated microglia and BAMs, in addition to their immunological role, are essential for the integrity of the BBB and control of its permeability through direct actions as well as by influencing vascular tone. The BBB itself is influenced by aging and is likely to be involved in PD pathogenesis. Aging-related changes of the BBB include a loss of pericyte and endothelial tight junctions, increasing the leakiness of peripheral cytokines and toxins. In addition, BECs upregulate VCAM1, which promotes the binding of circulating immune cells, inducing local inflammation. Furthermore, BECs lose their capacity for selective receptor-mediated transport of plasma proteins with age, which could negatively influence the microenvironment of dopaminergic neurons. The aging of the immune system itself is likely caused by the involution of the thymus with a reduction of naïve immune cells and an expansion of polyclonal antigen-experienced T cells triggered by recurrent life-long exposure to common pathogens (e.g., CMV). These senescent immune cells display a common senescent phenotype with enhanced secretion of pro-inflammatory cytokines sustaining a chronic low-grade inflammation (inflammaging). With age, the homeostatic T cell proliferation as a statistical process might as well lead to the selection of T cells with increased affinity to self- or neoantigens (as proposed in PD pathogenesis), facilitating autoimmune responses. Furthermore, regulatory inflammation-limiting and autotolerance-promoting populations of Tregs are diminished in relation to effector T cells. This figure represents a schematic overview and more detailed explanations and references can be found in the main body of the article. Created with BioRender.com. PD: Parkinson’s disease; ROS: reactive oxygen species; BAMs: border-associated macrophages; BBB: blood-brain barrier; BEC: brain endothelial cell; VCAM1: vascular cell adhesion molecule 1; CMV: cytomegalovirus; α-syn: α synuclein; CNS: central nervous system;
DOPAMINERGIC NEURONS, TIRELESS WORKERS VULNERABLE TO AGE?
The SN pars compacta (SNc) contains dopaminergic neurons or subsets of those neurons that seem to be especially susceptible to the effects of aging. Postmortem studies have found a reduced density of neurons in the SNc in presumably healthy-aged individuals and estimated the rate of neurodegeneration to be 5%-10% per decade[4-7]. This pronounced neuronal loss is in contrast to other brain regions such as the neocortex or the hippocampus which show much smaller reductions in healthy aging[8]. Several original articles and review articles have tried to shed light on the selective vulnerability of dopaminergic neurons in the SNc in healthy aging[1,9-13].
The main hypotheses are connected to the structure and/or the function of these neurons and have been recently reviewed extensively by others[9]. Structurally, the sheer size of the axonal arbor and the estimated number of 1-2 million transmitter release sites for a single neuron[13] might lead to a high bioenergetic demand[14] that induces mitochondrial oxidative stress and downstream neuronal dysfunction[15]. Functionally, the slow and tonic activity of dopaminergic SNc neurons in the absence of any stimulation is accompanied by strong changes in intracellular calcium concentrations. In combination with the low intracellular calcium buffering capacity of SNc dopaminergic neurons, this leads to periodically high free calcium concentrations, which in turn increase mitochondrial oxidative phosphorylation in a feedforward loop and cause oxidative stress[9,16]. The second hypothesis associated with function is connected to the neurotransmitter dopamine: metabolized and oxidized dopamine has been shown to damage different cellular components[17,18] and the successive buildup of neuromelanin has been linked to neurodegeneration and cellular dysfunction[19]. Based on these particular traits, SNc dopaminergic neurons have been described as “at the edge” of their energetic, proteolytic, and autophagic capacity, leaving little room to adapt to age-induced deteriorations while simultaneously requiring a higher turnover of damaged organelles and proteins compared to other neurons[9,10].
From this, it is readily conceivable that the general hallmarks of cellular aging[20] but especially mitochondrial dysfunction, oxidative damage, impaired molecular waste disposal, impaired DNA repair, and dysregulated calcium homeostasis (for a recent review, see also[21]) have stronger effects in SNc dopaminergic neurons. This hypothesis is supported by genetic mutations known to cause early-onset forms of PD or increase the risk of developing PD later in life, from which the great majority is connected to mitochondria or molecular waste disposal. PTEN-induced kinase 1 (PINK1), Parkinsonism-associated deglycase (PARK7), parkin RBR E3 ubiquitin protein ligase (PRKN), F-box only protein 7 (FBXO7), polymerase gamma (POLG), vacuolar protein sorting 13 homolog C (VPS13C), and HtrA serine peptidase 2 (HTRA2) are implicated in mitochondrial function, while ATPase cation transporting 13A2 (ATP13A2), glucosylceramidase beta (GBA), and leucine-rich repeat kinase 2 (LRRK2) are thought to play a role in both lysosomal and mitochondrial functioning[22]. Additionally, PTEN-induced kinase 1 (PINK1), parkin RBR E3 ubiquitin protein ligase (PRKN), endophilin A1 (EndoA1), and α synuclein (α-syn) pertain to autophagy as well[23]. Other mutations are indicated in synaptic vesicle formation, synaptic vesicle trafficking, and endosomal trafficking[22], all of which are most likely under high stress in the energy-demanding and highly branched dopaminergic axonal arbor. Furthermore, effective vesicle trafficking has been linked to mitochondrial quality control[24]. As the most common genetic risk factor for PD, GBA mutations have been extensively studied and highlight the complexity of the interactions between mitochondrial functioning and lysosomal autophagic pathways[25]. GBA encodes the lysosomal enzyme β-glucocerebrosidase (GCase), which converts glucosylceramide to ceramide and glucose. Certain mutations in this enzyme cause dysfunctions in the lysosomal autophagic pathway, but they have also been linked to mitochondrial defects and impaired mitochondrial function, most likely due to impaired mitophagy[26]. In a recent study, Baden
Recent preclinical work by González-Rodríguez et al. has, for the first time, demonstrated that the loss of function in mitochondrial complex I is sufficient to cause a levodopa-responsive form of Parkinsonism in a mouse model[28]. The authors selectively deleted the Ndufs2 (the gene coding for the catalytic subunit of mitochondrial complex I) in dopaminergic neurons and could show that an early loss of the dopaminergic phenotype in nigrostriatal axons that was accompanied by deficits in fine motor skills and motor learning. These results emphasize the importance of mitochondrial function and energy homeostasis in the pathophysiology of PD. The pivotal role of mitochondria in PD is further underpinned by preclinical findings that show improvements in PD following interventions that aim to improve mitochondrial function[29], biogenesis[30], or mitophagy[31] (for two recent and extensive reviews, see[32,33]). Especially striking in this context is the fact that stereotaxic, but also intravenous and intranasal transfer of allogenic mitochondria was associated with improvement in locomotor function and less degeneration of dopaminergic neurons in rodent models of PD[34-36]. Other non-pharmacological interventions like intermittent fasting and exercise that show promise in the treatment of PD have similarly been linked to enhanced mitochondrial biogenesis and the regulation of mitochondrial function[37-40].
The second hypothesis related to function is connected to the production and the successive processing of the neurotransmitter dopamine. Although the toxicity of dopamine has been put into question by the LEAP study[41] and the sub-Saharan Africa study[42] (i.e., long-term substitution of levodopa does not cause accelerated symptom progression), recent advances in creating neuromelanin accumulating animal models have put further emphasis on the pathophysiological role of neuromelanin in PD[19]. Neuromelanin is a dark pigment found only in a subset of catecholaminergic neurons such as the dopaminergic neurons of the SNc or the noradrenergic neurons of the locus coeruleus. It accumulates with advancing age and neurons seem to be unable to eliminate the pigment from the cell body[43]. As neuromelanin is most likely formed from dopamine or dopamine precursors through either oxidation or enzymatic catalysis with an unidentified tyrosinase, it is directly connected to dopaminergic function[44]. It has been shown that the overexpression of human tyrosinase in dopaminergic neurons leads to age-dependent neuromelanin accumulation, nigrostriatal degeneration, and a PD-like phenotype in rats, which could be alleviated by enhancing lysosomal proteostasis[45]. A more recent study in the same animal model found that the overexpression of vesicular monoamine transporter 2 (VMAT2) could prevent the buildup of neuromelanin by enhanced dopamine vesicular encapsulation resulting in the long-term preservation of motor function and neuronal integrity in the SNc[46]. Since the accumulation of neuromelanin is inherently linked to aging[43], it could present a second mechanism that connects aging and PD.
It is not fully clear how the stated factors influence each other, and a combination in the form of multiple hits has been proposed to cause excessive neuronal loss in PD[47]. The mechanisms elucidated so far, however, do not take into account the role of α-syn aggregation and Lewy pathology, which is thought to play a critical role in PD pathology backed up by genetical, preclinical, clinical and postmortem studies[48-52]. The link between these theories could be generated by selective age-induced vulnerability to the spread of a form of misfolded α-syn in SNc dopaminergic neurons. Since a hypothesized “external” triggering of α-syn misfolding, i.e., by a viral infection or toxins, as well as an “internal” triggering of α-syn misfolding, i.e., by oxidization or aberrant modification, are both statistical processes, they both become more likely with advanced age. Furthermore, the immunogenic potential of accumulated and misfolded α-syn[53,54] could create a chronic inflammatory state fueled by a positive feedback loop between neuronal death and immune cell stimulation. The role of the immune system in aging, neurodegeneration, and PD will be discussed in the following section.
THE AGING IMMUNE SYSTEM, A PERPETRATOR OF NEURODEGENERATION AND PD?
Over the last few years, the role of the immune system in the context of neurodegenerative diseases and their importance for the pathogenesis and maintenance of neurodegenerative processes have become a focus of research. In particular, the role of age-related changes in the functionality of the immune system, including maintained self-tolerance and anti-inflammatory potency, is debated to play a role[55,56]. Despite substantial research efforts, the majority of the immunological pathways linked to neurodegenerative diseases, such as PD or AD, are still poorly understood. While initially, research has focused on the role of the innate immune system, more recent studies investigated the role of peripheral immune cells and their ability to infiltrate the brain parenchyma. Age-related phenomena that hint towards a gradually failing immune system include a weakened response to novel antigens (e.g., influenza)[57], enhanced infection susceptibility[58], reduced effectiveness of vaccines[59], and age-related autoimmunity[56]. Today’s understanding of PD includes a multifactorial genesis involving a complex interplay of various factors, including genetic predisposition, age, immunological responses to bacterial and viral infection, and toxin exposure[60].
Immunosenescence and inflammaging constitute hallmarks of the aging immune system
Central terms in age-associated immune system alterations include “immunosenescence” and “inflammaging”[60,61].
Immunosenescence describes the depletion of the immune repertoire mainly by a decline in the naïve T cell pool with a concomitant accumulation of highly oligoclonal memory immune cells, especially terminally differentiated effector memory cells re-expressing CD45RA (TEMRA)[62]. These senescent T cells are antigen-experienced, providing “antigenic memory”, but have a diminished capacity to proliferate (“replicative senescence”[63]), and display a pro-inflammatory phenotype[64,65]. This shared senescence-associated secretory phenotype (SASP) is characterized by the production of pro-inflammatory cytokines, growth modulators, and chemotactic proteins, especially interleukin-6 (IL-6)[66]. Although these cytokines are also present during acute inflammation as a physiological response to tissue damage or pathogens[67], SASP-induced inflammation is long-lasting and therefore tissue-damaging[68]. This chronic low-grade inflammation that occurs with aging and senescence has been termed inflammaging.
Repeated lifelong exposure to novel antigens and the consequent specialization of T cells, coupled with gradual atrophy of the thymus with advancing age[56,69], is assumed to be the cause of the growing TEMRA population in older individuals. In addition, the infection with persistent viruses [specifically, cytomegalovirus (CMV)] has been implicated as a major driver of the accumulation of senescent T lymphocytes as these were significantly increased in the CMV-seropositive elderly population[70-73].
The constantly shrinking number of naïve T cells capable of responding to novel antigens includes a loss of naïve-like regulatory T cells (Tregs) that are able to limit inflammation and maintain self-tolerance.
Evidence for the immunological involvement in PD pathogenesis
The analysis of postmortem human PD brains provided the first evidence for the involvement of T cells by discovering CD8 and CD4 T cells, either neighboring blood vessels or adjacent to dopaminergic neurons[74]. Using α-syn overexpressing murine models, early CD4 and CD8 T cell infiltration could be demonstrated as a pathognomonic mechanism while T cells also amplified the amount of α-syn aggregates by triggering a pro-inflammatory phenotype in central nervous system (CNS) myeloid cells[75-77]. The primarily CD4-mediated dopaminergic toxicity was also validated in a neurotoxin-based mouse model[78], as well as through in vitro data[79]. Additionally, again in an α-syn overexpression model, the knockout of the T cell receptor, as well as CD4, reduced the major histocompatibility complex (MHC) II-mediated myeloid cell response to α-syn, preventing neuronal loss, while the deletion of CD8 did not[77].
A neuropathological study that assessed T cell infiltration in the SN throughout different PD stages (defined by the anatomical distribution of α-syn aggregates)[80] found that CD8 T cell infiltration was most pronounced in the earliest stage of PD when no protein aggregates or dopaminergic neuron loss were present yet. This suggests that CD8-mediated attacks may initiate neuronal death and α-syn aggregation and may abate with the progression of neurodegeneration[80].
In 2017, Sulzer et al. found that peripheral T cells from PD patients’ blood were specific for α-syn derived epitopes (Y39, S129)[54]. Interestingly, a follow-up study reported that this α-syn-specific T cell activation was also most pronounced in early-stage PD fading with progressing neurodegeneration[81]. It is likely that the T cell involvement described above is also reflected in the alterations of peripheral T cell populations through crosstalk of central and peripheral compartments, whereby a disrupted BBB (see below) may contribute.
Limited research has been dedicated to examining age-related alterations in the adaptive immune system in the specific context of PD. However, a recent comprehensive review has addressed this subject, prompting only a brief discussion of the major findings[82]. As mentioned above, aging affects CD4 T cells later and less than CD8 T cells[83]. While there is some opposing evidence considering immunosenescence in PD, the majority of studies investigating lymphocyte populations suggested that naïve CD8 T cells were significantly decreased in the peripheral blood of PD patients, whereas pro-inflammatory CD8 T cells were increased[84-86]. However, another study found a reduction in CD8 TEMRA cells and a lower expression of the cell-aging marker p16, suggesting less senescent CD8 T cells in PD[87]. It is of note, however, that no association between T cell senescence and measures of disease progression has been observed (e.g., severity of motor or cognitive symptoms), but reduced TEMRA counts have been observed across all disease stages including newly diagnosed cases[87,88]. This suggests that immunosenescence might play a role in disease pathogenesis rather than in progression. As mentioned above, chronic viral infections (mainly CMV) have been identified as the main drivers for CD8 T cell senescence. Interestingly, specifically in CMV seropositive PD patients, the TEMRA phenotype appeared to be attenuated, suggesting intrinsic differences in the T cell response to chronic infections in PD vs. controls[89].
Considering compositional shifts in lymphocyte subpopulations, again, conflicting results have been reported. A meta-analysis including almost 1,000 PD patients and age-matched controls each found a significant reduction in the CD4:CD8 T cell ratio[85], while a study with a total of 1,500 paticipants including PD patients with age- and gender-matched healthy controls found the opposite[90]. Analyzing CD4 phenotypes in-depth, Chen et al. observed an increased proportion of circulating pro-inflammatory IFN-γ-releasing Th1 and Th17 cells and a decreased number of anti-inflammatory regulatory T cells in the peripheral blood of PD patients[91]. The detrimental role of pro-inflammatory Th1/Th17 cells was further shown in a neurotoxin-based (MPTP) model of PD where naïve CD4 T cells exposed to nitrated α-syn developed Th1/Th17 phenotype, which in turn led to an exacerbated MPTP-induced dopaminergic cell death[78].
On the other hand, immune mechanisms responsible for governing and modulating chronic inflammation appear to be compromised PD. Specifically, PD patients exhibit a significant reduction in Tregs[92]. A potential role of Treg insufficiency in disease development and progression is further implicated by mitigating neurodegenerative processes and clinical deterioration by restoring the quantity and functionality of Tregs. This has been shown preclinically[78,93] and has been tested for safety and response in immune biomarkers in patients with PD[94] using the granulocyte-macrophage colony-stimulating factor (GM-CSF) sargramostim, which is known to shift pro-inflammatory effector T cells (Teff) to Tregs[95-97].
The T cell response might also be affected by changes in antigen-presenting cells, which play a critical role in T cell priming. In the brain, microglia serve as the tissue-resident immune cells that constantly survey the environment[98]. Evidence of microglial activation in PD stems from neuropathological studies of the SN of postmortem brains[99,100], positron emission tomography (PET) studies in PD patients at different disease stages[101-103], and preclinical data using toxin-based as well as α-syn overexpressing mouse models of PD[75,104,105]. In addition, a specific isotype of the human leukocyte antigen gene (HLA-DR) has been identified by a genome-wide association study (GWAS) as a genetic risk factor for late-onset PD[106]. HLA is part of the antigen-presenting machinery used to activate T cells and is specifically expressed in microglia within the brain[107]. Furthermore, it was shown that the deliberate induction of microgliosis by stereotactic injection of the bacterial toxin lipopolysaccharide (LPS) into the mouse brain detrimentally affects dopaminergic neurons in the substantia nigra, contributing to their degeneration[108,109].
With age, microglia also undergo senescence. Morphologically, they display increased fragmentation and gnarling in their processes, and functionally, they exhibit reduced phagocytic and migratory capabilities[110]. Both activated and senescent microglia produce inflammatory cytokines and may primarily arise from inflammatory insults[111,112]. In PD, microglial activation is thought to be a significant part of the disease process integrated either as a cause or consequence[113,114]. While microglial phagocytosis of extracellular misfolded α-syn can lead to its degradation and clearance[115], microglia can also release α-syn through exosomes, promoting its spread[116] - indicating a complex role in both clearing and transferring α-syn pathology. A recent study investigated the impact of aging and glial senescence on α-syn clearance, revealing that aged mice and microglia exhibit compromised clearance capacity, primarily linked to dysfunction in the autophagy-lysosome system, shedding more light on the role of senescent glia in PD pathogenesis[117]. Taken together, these observations indicate a pivotal role for microglia in PD disease progression and raise interest in studying their functional roles during PD pathology.
For a relatively long time, microglia have been understood to be the main players in the innate immune system of the CNS, and only recently has a second category of macrophages come to the attention of researchers. The so-called border-associated macrophages (BAMs) are not located in the brain parenchyma but in the adjacent spaces between the meninges, the choroid plexus, and perivascular spaces. Microglia and BAMs share common progenitors but differ in morphology and function[118]. With their unique localization between the brain parenchyma and peripheral tissues, BAMs have a specific importance in the defense against pathogens. In addition, their proximity to the vascular space allows them to regulate the permeability of circulating cells and mediators by influencing vascular contractility and BBB permeability (see below).
A recent study by Schonhoff et al. found that BAMs, in contrast to parenchymal microglia, are essential for CD4-mediated inflammation and consequent dopaminergic degeneration in an α-syn overexpressing model of PD[119]. Furthermore, in the neuropathological analysis of postmortem brains, significantly higher numbers of CD3-positive lymphocytes were found in the midbrain area in the immediate vicinity of BAMs in PD patients in comparison to healthy controls[119], which in turn suggests the maintenance of inflammation through crosstalk with the peripheral circulating immune cells. This observation supports a Parkinsonian immune-mediated pathogenesis, but a specific age-mediated change in the functionality of BAMs should be further investigated.
Interestingly, although initially of the same embryonic origin (yolk sac)[120], with age, dying macrophages can be derived from monocytes recruited from the periphery which are derived from hematopoietic stem cells[121]. This dual origin and composition of brain macrophages is virtually absent in the young brain and the proportion of monocyte-derived macrophages increases with age. These are further characterized by an increased pro-inflammatory profile and circulating monocytes themselves display altered immune phenotypes with age[122].
Inflammaging
When cells are damaged, they release different kinds of danger signals to the extracellular space, which promote inflammatory responses by binding to specific pattern-recognition receptors. Danger molecules are broadly categorized into non-self and self, also termed pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs), respectively. A third category of so-called quasi-self molecules includes products from gut microbiota. This large variety of stimuli converges on the same evolutionarily preserved receptors to trigger inflammatory responses[123] Age-related accumulation of such events causes a gradual rise in the basal level of inflammation that may last for decades, eventually resulting in inflammaging.
In PD, a large body of work has demonstrated higher levels of peripheral pro-inflammatory mediators. A recent meta-analysis (encompassing 25 studies with a total of 1,547 PD patients and 1,107 controls) revealed significantly elevated concentrations of CRP, tumor necrosis factor α (TNF-α), IL-1β, IL-6, CRP, IL-10, and IL-2 in the serum of PD patients versus controls[124]. A moderate but significant increase in TNF-α, IL-1β,
Demented patients with PD show elevated cytokine levels (CRP, IL-6, IL-8) in their CSF compared to cognitively unaffected individuals, and higher levels of CRP have been associated with cognitive impairment, depression, and fatigue[126].
Currently, it is not fully understood if the mentioned phenomena are caused by a dysregulation of the immune system due to aging or by disease-related processes. It has recently become known that α-syn aggregates are detectable in the peripheral blood of patients with PD and can activate Toll-like receptors, thereby causing an inflammatory response[54] and the induction of α-syn-specific T cells[127,128] A recent study further investigated antigen-specific T cell responses directed towards commonly encountered human pathogens and vaccines (viral and bacterial, e.g., influenza, rhinovirus, tetanus) in patients with PD and age-matched healthy controls[129]. It was found that PD patients and healthy controls showed similar T cell activation levels and levels of cytokines produced upon presentation of these common pathogen-derived antigens. These results, for the first time, suggest that T cell dysfunction and enhanced inflammatory T cell responses in PD may be more directed towards autoantigens like α-syn rather than common foreign antigens. However, the development of this kind of autoimmune response, in turn, may be facilitated by the aging immune system with the above-mentioned processes (depletion of naïve T cell populations and shift from auto-tolerogenic Tregs towards pro-inflammatory Teffs).
Because of the increased production of pro-inflammatory cytokines, the theory that anti-inflammatory treatments will be beneficial for diseases of aging has arisen. Epidemiologic studies found that people with regular use of non-aspirin non-steroidal anti-inflammatory drugs (NSAIDs, e.g., ibuprofen) had a significantly lower risk of developing PD relative to non-regular users[130]. Preclinical research further supported the protective role of NSAIDs in PD, showing that ibuprofen protected dopaminergic neurons against toxicity in vitro and against MPTP-induced neurotoxicity in rodents[131].
While it is acknowledged that the aforementioned processes exhibit sex-dependent differences, the focus of this review on aging precludes a detailed discussion of this aspect. For a comprehensive understanding of sex differences in aging, it is recommended to refer to recently published insightful reviews. The first review provides an overview of sex differences in aging, encompassing various physiological aspects[132]. For a more specific exploration of sex differences in neurodegenerative diseases, particularly PD, the review by Gillies
THE BLOOD-BRAIN BARRIER, (MORE THAN) A WATCHMAN BLINDED BY AGE?
The term BBB describes the interface between the circulating blood and the extracellular fluid of the CNS. Classically, it is understood to comprise brain endothelial cells (BECs), pericytes, astrocytic endfeet, and extracellular matrix that tightly regulate the exchange of proteins, metabolites, ions, and cells. However, more recent work has shown that microglial cells play a crucial role in BBB function in health and disease[134] and that BAMs could contribute to the barrier as well[135]. In PD, BBB dysfunction was found in humans as well as in different animal models[136-139]. Since the BBB is known to control the exchange of biological substances and to limit immune cell migration from the periphery, it is commonly regarded as essential for maintaining the homeostasis of the microenvironment in the brain[140,141]. It is currently not fully clear whether BBB dysfunction is an early event in PD pathophysiology or happens further downstream in the disease progression. Murine studies found that BBB disruption can be initiated by the common neurotoxins 6-OHDA and MPTP as well as overexpression of α-syn[138,142,143]. However, it is also conceivable that BBB dysfunction is an earlier process in the pathophysiological cascade in PD as BBB disruption through stereotactic VEGF injection selectively damaged dopaminergic neurons but not GABAergic neurons[137]. Clinical studies found an association between cognitive dysfunction and gait impairments with vascular comorbidity in people with PD[144,145], possibly associated with BBB dysfunction. Regardless of the timing of BBB dysfunction in PD, it has been shown that aging increases BBB leakage of contrast agents[146,147], changes its transporter profiles[148-151], alters the extracellular matrix composition[152,153], and is associated with the loss of pericytes[154,155] (as recently reviewed extensively elsewhere[156]).
There is only sparse data on the precise effects of aging on the cell types that make up the BBB. Except for the aging-associated loss of pericytes, Nikolakopoulou et al. demonstrated in an inducible knockout model that pericyte loss results in neurodegeneration (within days) and behavioral deficits (within two weeks) in mice[157]. In the context of astrocytes, aging has been found to increase the number of reactive astrocytes that express genes associated with neuroinflammation and enhance oxidative metabolism in astrocytes[158,159]. Furthermore, reactive astrocytosis has been shown to impair Sonic hedgehog signaling, which is an important regulator of BBB integrity[160]. However, the impact of age-associated changes on astrocytic endfeet in the context of the BBB has not been studied. Recent studies have additionally investigated the role of aging BECs more closely and found that they display elevated markers for inflammation and stress response[161-163]. In an elegant study, Yousef et al. could show that the plasma of aged mice activated microglial cells via vascular cell adhesion molecule 1 (VCAM1) in BECs of young mice[163]. The authors found no leukocyte recruitment to the brain and concluded that leukocytes tether to VCAM1 without crossing the barrier and sustain an inflammatory reaction by secreting pro-inflammatory factors in the immediate vicinity of BECs. This inflammatory reaction is then relayed to the brain microenvironment and microglia via BECs. It is important to note that the mouse model used did not have T, B, or NK cells. Other studies using models that were not depleted for T and B cells, on the other hand, were able to show increased immune cell infiltration to the brain (T cells) or meninges (B cells) in aging mice[164,165] but not in humans[166]. Apart from the pro-inflammatory phenotype, BECs also have a significantly diminished ability to transport plasma proteins to the brain as they age[167]. This surprising finding, which is in contrast to tracer studies, was demonstrated by Yang et al. with a new labeling approach for the plasma proteome. In the same study, the loss in protein transport was linked to an age-associated shift from receptor-mediated transcytosis to caveolar transcytosis as a less specific mechanism[167]. These perspectives challenge the classical view of the aging BBB as a passive structure and put emphasis on aging-associated alterations in active transport functions. Against the background of the selective vulnerability of SNc dopaminergic neurons, these changes could play a role in aging-associated degeneration by changing the local microenvironment and disrupting homeostasis. Interestingly, this could be a bidirectional relationship since oxidative stress can in turn cause dysfunction of the BBB and influence astrocytes, pericytes, and BECs[168].
In recent years, it has been shown that microglial cells contribute to BBB repair as well as integrity and can influence vascular tone[169-171]. Interestingly, microglia can promote BBB integrity via Claudin-5 expression following acute inflammatory events but have also been shown to cause BBB leakage through phagocytosis of astrocytic endfeet in chronic inflammatory conditions[171]. Similarly, activated microglia can induce the loss of pericytes via TGF-β signaling in models of Alzheimer’s disease[172] and have been shown to cause BBB disruption via activation of MMP-2/-9[173] in a mouse model of PD. The role of microglial interactions with cells that form the BBB is further underpinned by the recent description of capillary-associated microglia (CAM) that reside in close proximity to microvessels and could make up around 30% of the microglial population in the mouse brain. Although acute depletion of these cells did not increase BBB permeability, they were demonstrated to regulate vascular tone in a PANX1-P2RY12-dependent manner[170]. In another study, inhibition or deletion of the P2RY12 receptor in microglia was found to prolong BBB closure after acute laser ablation[174]. Another important type of cells that interact with the BBB are perivascular macrophages (PVMs), a subtype of BAMs (see previous chapter), and although their main role is considered to be in immune surveillance and autophagy, studies also point to a modulation of vascular tone and BBB permeability[118]. Like in microglial cells, there seems to be a dual role in the interaction with the BBB, where PVMs maintain and regulate BBB function in health, but can cause BBB damage and leakage in disease or inflammatory conditions[135,175]. PVMs are considered highly stable and possess self-renewal capacity under physiological conditions[176]. Interestingly, PVMs can be replenished by infiltrating monocytes after insults or in pathological states[135]. Not much is known about the specific effects of aging on PVMs in the context of their interaction with the BBB, but recent work suggests that their number increases in older humans[177] and that they lose functions associated with cerebrospinal fluid regulation[178]. Furthermore, another subtype of BAMs, meningeal macrophages, show an increase in MHC-II+ dural macrophages with aging, while MHC-II- dural macrophages diminish[179], possibly making them more likely to activate T cells and promote inflammation. These observations make it conceivable that the ability of PVMs to maintain and repair the BBB also diminishes with age and that they shift to more pro-inflammatory phenotypes, thereby actively increasing the permeability of the BBB.
Importantly, there is also substantial crosstalk between the BBB and immune cells in the periphery[180,181]. High levels of inflammatory cytokines such as IL-6, TNF-α or IL-1β in the blood (see previous paragraphs for association with PD) can cause decreased expression or breakdown of essential tight junction proteins like Occludin and Claudin-5, resulting in a more permissive BBB[182,183]. In addition, peripheral inflammation has been linked to the upregulation of cellular adhesion molecules (e.g., VCAM1, ICAM1, and E-Selectin) on BECs, which in turn facilitate the entry of peripheral immune cells into the CNS[182], potentially causing a feedforward loop of dysregulated immune function and disruption of the BBB. The relevance of these interactions in PD, however, is only poorly understood and studies establishing causality or a timeline of pathophysiological events are currently missing.
Establishing the precise role of the BBB in the context of aging, inflammation, and neurodegeneration is increasingly imperative, as focused-ultrasound-induced BBB opening has become more available in recent years as a tool for drug delivery[184]. The technique induces a transient, focal, and reversible opening of the BBB through the interaction of a focused ultrasound beam with systemically administered microbubbles[185]. The procedure has been shown to be safe in people with Alzheimer’s disease[186] and PD[187,188] and various animal models[184]. From this, it would be conceivable that short openings of the BBB can safely be tolerated, while chronic long-term openings result in immune infiltration and neurodegeneration. However, long-term data are missing due to the novelty of the procedure. Apart from drug delivery, the ability of the technique for localized and transient BBB opening could also help in better understanding the role of the BBB in neurodegenerative diseases and aging.
CONCLUSION
The selective vulnerability of dopaminergic neurons in the substantia nigra pars compacta to the effects of aging is a multimodal phenomenon, but recent advances in animal models of PD have established a central pathophysiological role of oxidative stress, mitochondrial dysfunction, and neuromelanin accumulation. Despite these advances, there is currently no comprehensive theory that unites selective vulnerability with other factors implicated in the pathophysiology of PD, such as Lewy pathology, the response of the immune system, and BBB disruption. We have laid out in this review that age-associated changes in the immune system (i.e., lower anti-inflammatory potency and lower self-tolerance) and the BBB (i.e., impaired homeostasis, pro-inflammatory phenotype) can contribute to pathophysiological changes in PD, making the effects of aging a multisystem phenomenon. This review did not touch on the possibly important contributions of aging in glial cells, the glymphatic system, or the microbiome and further studies will be needed to establish a comprehensive pathophysiological cascade and possible causality. Nevertheless, given the current evidence, it seems likely that multimodal effects of aging on the levels of SNc dopaminergic neurons, the immune system, and the BBB increase the likelihood of excessive neurodegeneration and, subsequently, PD. Furthermore, this multisystem approach allows for multiple angles to potentially initiate pathophysiological processes which could partially explain the heterogeneity in PD.
DECLARATIONS
Authors’ contributions
Study conception and design: Bendig J, Frank A, Reichmann H
Literature search and review: Bendig J, Frank A
Draft manuscript and preparation: Bendig J, Frank A, Reichmann H
Availability of data and materials
Not applicable.
Financial support and sponsorship
Not applicable.
Conflicts of interest
All authors declared that there are no conflicts of interest. Reichmann H is an Associate Editor of the journal Ageing and Neurodegenerative Diseases.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2024.
REFERENCES
1. Coleman C, Martin I. Unraveling Parkinson’s disease neurodegeneration: does aging hold the clues? J Parkinsons Dis 2022;12:2321-38.
2. de Rijk MC, Breteler MM, Graveland GA, et al. Prevalence of Parkinson’s disease in the elderly: the Rotterdam Study. Neurology 1995;45:2143-6.
3. Kontis V, Bennett JE, Mathers CD, Li G, Foreman K, Ezzati M. Future life expectancy in 35 industrialised countries: projections with a Bayesian model ensemble. Lancet 2017;389:1323-35.
4. Rudow G, O’Brien R, Savonenko AV, et al. Morphometry of the human substantia nigra in ageing and Parkinson’s disease. Acta Neuropathol 2008;115:461-70.
5. Fearnley JM, Lees AJ. Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain 1991;114:2283-301.
6. Ma SY, Röytt M, Collan Y, Rinne JO. Unbiased morphometrical measurements show loss of pigmented nigral neurones with ageing. Neuropathol Appl Neurobiol 1999;25:394-9.
7. Buchman AS, Shulman JM, Nag S, et al. Nigral pathology and parkinsonian signs in elders without Parkinson disease. Ann Neurol 2012;71:258-66.
9. Gonzalez-Rodriguez P, Zampese E, Surmeier DJ. Disease mechanisms as subtypes: mitochondrial and bioenergetic dysfunction. Handb Clin Neurol 2023;193:53-66.
10. Gonzalez-Rodriguez P, Zampese E, Surmeier DJ. Selective neuronal vulnerability in Parkinson’s disease. Prog Brain Res 2020;252:61-89.
11. Surmeier DJ, Obeso JA, Halliday GM. Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci 2017;18:101-13.
12. Pandya VA, Patani R. Region-specific vulnerability in neurodegeneration: lessons from normal ageing. Ageing Res Rev 2021;67:101311.
13. Bolam JP, Pissadaki EK. Living on the edge with too many mouths to feed: why dopamine neurons die. Mov Disord 2012;27:1478-83.
14. Pissadaki EK, Bolam JP. The energy cost of action potential propagation in dopamine neurons: clues to susceptibility in Parkinson’s disease. Front Comput Neurosci 2013;7:13.
15. Kowalczyk P, Sulejczak D, Kleczkowska P, et al. Mitochondrial oxidative stress - a causative factor and therapeutic target in many diseases. Int J Mol Sci 2021;22:13384.
16. Guzman JN, Ilijic E, Yang B, et al. Systemic isradipine treatment diminishes calcium-dependent mitochondrial oxidant stress. J Clin Invest 2018;128:2266-80.
17. Segura-Aguilar J, Paris I, Muñoz P, Ferrari E, Zecca L, Zucca FA. Protective and toxic roles of dopamine in Parkinson’s disease. J Neurochem 2014;129:898-915.
18. Burbulla LF, Song P, Mazzulli JR, et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 2017;357:1255-61.
19. Vila M. Neuromelanin, aging, and neuronal vulnerability in Parkinson’s disease. Mov Disord 2019;34:1440-51.
20. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell 2013;153:1194-217.
21. Mattson MP, Arumugam TV. Hallmarks of brain aging: adaptive and pathological modification by metabolic states. Cell Metab 2018;27:1176-99.
22. Day JO, Mullin S. The genetics of Parkinson’s disease and implications for clinical practice. Genes 2021;12:1006.
23. Lynch-Day MA, Mao K, Wang K, Zhao M, Klionsky DJ. The role of autophagy in Parkinson’s disease. Cold Spring Harb Perspect Med 2012;2:a009357.
24. Sugiura A, McLelland GL, Fon EA, McBride HM. A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J 2014;33:2142-56.
25. Smith L, Schapira AHV. GBA variants and Parkinson disease: mechanisms and treatments. Cells 2022;11:1261.
26. Zheng W, Fan D. Glucocerebrosidase mutations cause mitochondrial and lysosomal dysfunction in Parkinson’s disease: pathogenesis and therapeutic implications. Front Aging Neurosci 2022;14:851135.
27. Baden P, Perez MJ, Raji H, et al. Glucocerebrosidase is imported into mitochondria and preserves complex I integrity and energy metabolism. Nat Commun 2023;14:1930.
28. González-Rodríguez P, Zampese E, Stout KA, et al. Disruption of mitochondrial complex I induces progressive parkinsonism. Nature 2021;599:650-6.
29. Que R, Zheng J, Chang Z, et al. Dl-3-n-butylphthalide rescues dopaminergic neurons in Parkinson’s disease models by inhibiting the NLRP3 inflammasome and ameliorating mitochondrial impairment. Front Immunol 2021;12:794770.
30. Kim KH. Intranasal delivery of mitochondrial protein humanin rescues cell death and promotes mitochondrial function in Parkinson’s disease. Theranostics 2023;13:3330-45.
31. Lin MW, Lin CC, Chen YH, Yang HB, Hung SY. Celastrol inhibits dopaminergic neuronal death of Parkinson’s disease through activating mitophagy. Antioxidants 2020;9:37.
32. Gao XY, Yang T, Gu Y, Sun XH. Mitochondrial dysfunction in Parkinson’s disease: from mechanistic insights to therapy. Front Aging Neurosci 2022;14:885500.
33. Prasuhn J, Davis RL, Kumar KR. Targeting mitochondrial impairment in Parkinson’s disease: challenges and opportunities. Front Cell Dev Biol 2021;8:615461.
34. Chang JC, Wu SL, Liu KH, et al. Allogeneic/xenogeneic transplantation of peptide-labeled mitochondria in Parkinson’s disease: restoration of mitochondria functions and attenuation of 6-hydroxydopamine-induced neurotoxicity. Transl Res 2016;170:40-56.e3.
35. Chang JC, Chao YC, Chang HS, et al. Intranasal delivery of mitochondria for treatment of Parkinson’s disease model rats lesioned with 6-hydroxydopamine. Sci Rep 2021;11:10597.
36. Shi X, Zhao M, Fu C, Fu A. Intravenous administration of mitochondria for treating experimental Parkinson’s disease. Mitochondrion 2017;34:91-100.
37. Lai JH, Chen KY, Wu JCC, et al. Voluntary exercise delays progressive deterioration of markers of metabolism and behavior in a mouse model of Parkinson’s disease. Brain Res 2019;1720:146301.
38. Zhao Y, Jia M, Chen W, Liu Z. The neuroprotective effects of intermittent fasting on brain aging and neurodegenerative diseases via regulating mitochondrial function. Free Radic Biol Med 2022;182:206-18.
39. Li J, Xu Y, Liu T, Xu Y, Zhao X, Wei J. The role of exercise in maintaining mitochondrial proteostasis in Parkinson’s disease. Int J Mol Sci 2023;24:7994.
40. Falkenburger B, Bernhardt N, Szegö E. Intermittent fasting reduces alpha-synuclein pathology and functional decline in mice. Research Square. [Preprint] July 5, 2023. [accessed on 2024 Mar 19]. Available from: https://www.researchsquare.com/article/rs-3077779/v1.
41. Verschuur CVM, Suwijn SR, Boel JA, et al. Randomized delayed-start trial of levodopa in Parkinson’s disease. N Engl J Med 2019;380:315-24.
42. Cilia R, Akpalu A, Sarfo FS, et al. The modern pre-levodopa era of Parkinson’s disease: insights into motor complications from sub-Saharan Africa. Brain 2014;137:2731-42.
43. Fedorow H, Halliday GM, Rickert CH, Gerlach M, Piederer P, Double KL. Evidence for specific phases in the development of human neuromelanin. Neurobiol Aging 2006;27:506-12.
44. Nagatsu T, Nakashima A, Watanabe H, Ito S, Wakamatsu K. Neuromelanin in Parkinson’s disease: tyrosine hydroxylase and tyrosinase. Int J Mol Sci 2022;23:4176.
45. Carballo-Carbajal I, Laguna A, Romero-Giménez J, et al. Brain tyrosinase overexpression implicates age-dependent neuromelanin production in Parkinson’s disease pathogenesis. Nat Commun 2019;10:973.
46. Gonzalez-Sepulveda M, Compte J, Cuadros T, et al. In vivo reduction of age-dependent neuromelanin accumulation mitigates features of Parkinson’s disease. Brain 2023;146:1040-52.
47. Sulzer D. Multiple hit hypotheses for dopamine neuron loss in Parkinson’s disease. Trends Neurosci 2007;30:244-50.
48. Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997;276:2045-7.
49. Gómez-Benito M, Granado N, García-Sanz P, Michel A, Dumoulin M, Moratalla R. Modeling Parkinson’s disease with the alpha-synuclein protein. Front Pharmacol 2020;11:356.
50. Siderowf A, Concha-Marambio L, Lafontant DE, et al. Assessment of heterogeneity among participants in the Parkinson’s Progression Markers Initiative cohort using α-synuclein seed amplification: a cross-sectional study. Lancet Neurol 2023;22:407-17.
51. Braak H, Del Tredici K, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 2003;24:197-211.
52. Angot E, Steiner JA, Lema Tomé CM, et al. Alpha-synuclein cell-to-cell transfer and seeding in grafted dopaminergic neurons in vivo. PLoS One 2012;7:e39465.
53. Reish HE, Standaert DG. Role of α-synuclein in inducing innate and adaptive immunity in Parkinson disease. J Parkinsons Dis 2015;5:1-19.
54. Sulzer D, Alcalay RN, Garretti F, et al. T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature 2017;546:656-61.
57. Thompson WW, Shay DK, Weintraub E, et al. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA 2003;289:179-86.
59. Goodwin K, Viboud C, Simonsen L. Antibody response to influenza vaccination in the elderly: a quantitative review. Vaccine 2006;24:1159-69.
60. Tansey MG, Wallings RL, Houser MC, Herrick MK, Keating CE, Joers V. Inflammation and immune dysfunction in Parkinson disease. Nat Rev Immunol 2022;22:657-73.
61. Fulop T, Larbi A, Dupuis G, et al. Immunosenescence and inflamm-aging as two sides of the same coin: friends or foes? Front Immunol 2017;8:1960.
62. Karrer U, Sierro S, Wagner M, et al. Memory inflation: continuous accumulation of antiviral CD8+ T cells over time. J Immunol 2003;170:2022-9.
63. Chou JP, Effros RB. T cell replicative senescence in human aging. Curr Pharm Des 2013;19:1680-98.
64. Akbar AN, Henson SM, Lanna A. Senescence of T lymphocytes: implications for enhancing human immunity. Trends Immunol 2016;37:866-76.
65. Geginat J, Lanzavecchia A, Sallusto F. Proliferation and differentiation potential of human CD8+ memory T-cell subsets in response to antigen or homeostatic cytokines. Blood 2003;101:4260-6.
66. Lee KA, Flores RR, Jang IH, Saathoff A, Robbins PD. Immune senescence, immunosenescence and aging. Front Aging 2022;3:900028.
67. Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 2010;5:99-118.
70. van der Heiden M, van Zelm MC, Bartol SJW, et al. Differential effects of cytomegalovirus carriage on the immune phenotype of middle-aged males and females. Sci Rep 2016;6:26892.
71. Derhovanessian E, Maier AB, Hähnel K, et al. Infection with cytomegalovirus but not herpes simplex virus induces the accumulation of late-differentiated CD4+ and CD8+ T-cells in humans. J Gen Virol 2011;92:2746-56.
72. Khan N, Shariff N, Cobbold M, et al. Cytomegalovirus seropositivity drives the CD8 T cell repertoire toward greater clonality in healthy elderly individuals. J Immunol 2002;169:1984-92.
73. Hadrup SR, Strindhall J, Køllgaard T, et al. Longitudinal studies of clonally expanded CD8 T cells reveal a repertoire shrinkage predicting mortality and an increased number of dysfunctional cytomegalovirus-specific T cells in the very elderly. J Immunol 2006;176:2645-53.
74. Brochard V, Combadière B, Prigent A, et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest 2009;119:182-92.
75. Sanchez-Guajardo V, Febbraro F, Kirik D, Romero-Ramos M. Microglia acquire distinct activation profiles depending on the degree of alpha-synuclein neuropathology in a rAAV based model of Parkinson’s disease. PLoS One 2010;5:e8784.
76. Sommer A, Fadler T, Dorfmeister E, et al. Infiltrating T lymphocytes reduce myeloid phagocytosis activity in synucleinopathy model. J Neuroinflammation 2016;13:174.
77. Williams GP, Schonhoff AM, Jurkuvenaite A, Gallups NJ, Standaert DG, Harms AS. CD4 T cells mediate brain inflammation and neurodegeneration in a mouse model of Parkinson’s disease. Brain 2021;144:2047-59.
78. Reynolds AD, Stone DK, Hutter JAL, Benner EJ, Mosley RL, Gendelman HE. Regulatory T cells attenuate Th17 cell-mediated nigrostriatal dopaminergic neurodegeneration in a model of Parkinson’s disease. J Immunol 2010;184:2261-71.
79. Sommer A, Marxreiter F, Krach F, et al. Th17 lymphocytes induce neuronal cell death in a human iPSC-based model of Parkinson’s disease. Cell Stem Cell 2018;23:123-31.e6.
80. Galiano-Landeira J, Torra A, Vila M, Bové J. CD8 T cell nigral infiltration precedes synucleinopathy in early stages of Parkinson’s disease. Brain 2020;143:3717-33.
81. Lindestam Arlehamn CS, Dhanwani R, Pham J, et al. α-Synuclein-specific T cell reactivity is associated with preclinical and early Parkinson’s disease. Nat Commun 2020;11:1875.
82. Kouli A, Williams-Gray CH. Age-related adaptive immune changes in Parkinson’s disease. J Parkinsons Dis 2022;12:S93-104.
83. Moro-García MA, Alonso-Arias R, López-Larrea C. When aging reaches CD4+ T-cells: phenotypic and functional changes. Front Immunol 2013;4:107.
84. Yan Z, Yang W, Wei H, et al. Dysregulation of the adaptive immune system in patients with early-stage Parkinson disease. Neurol Neuroimmunol Neuroinflamm 2021;8:e1036.
85. Baba Y, Kuroiwa A, Uitti RJ, Wszolek ZK, Yamada T. Alterations of T-lymphocyte populations in Parkinson disease. Parkinsonism Relat Disord 2005;11:493-8.
86. Wang P, Yao L, Luo M, et al. Single-cell transcriptome and TCR profiling reveal activated and expanded T cell populations in Parkinson’s disease. Cell Discov 2021;7:52.
87. Kouli A, Jensen M, Papastavrou V, et al. T lymphocyte senescence is attenuated in Parkinson’s disease. J Neuroinflammation 2021;18:228.
88. Williams-Gray CH, Wijeyekoon RS, Scott KM, Hayat S, Barker RA, Jones JL. Abnormalities of age-related T cell senescence in Parkinson’s disease. J Neuroinflammation 2018;15:166.
89. Vavilova JD, Boyko AA, Ponomareva NV, et al. Reduced immunosenescence of peripheral blood T cells in Parkinson’s disease with CMV infection background. Int J Mol Sci 2021;22:13119.
90. Chen X, Feng W, Ou R, et al. Evidence for peripheral immune activation in Parkinson’s disease. Front Aging Neurosci 2021;13:617370.
91. Chen Y, Qi B, Xu W, et al. Clinical correlation of peripheral CD4+-cell sub-sets, their imbalance and Parkinson’s disease. Mol Med Rep 2015;12:6105-11.
92. Saunders JAH, Estes KA, Kosloski LM, et al. CD4+ regulatory and effector/memory T cell subsets profile motor dysfunction in Parkinson’s disease. J Neuroimmune Pharmacol 2012;7:927-38.
93. Markovic M, Yeapuri P, Namminga KL, et al. Interleukin-2 expands neuroprotective regulatory T cells in Parkinson’s disease. NeuroImmune Pharm Ther 2022;1:43-50.
94. Olson KE, Abdelmoaty MM, Namminga KL, et al. An open-label multiyear study of sargramostim-treated Parkinson’s disease patients examining drug safety, tolerability, and immune biomarkers from limited case numbers. Transl Neurodegener 2023;12:26.
95. Olson KE, Namminga KL, Lu Y, et al. Granulocyte-macrophage colony-stimulating factor mRNA and neuroprotective immunity in Parkinson’s disease. Biomaterials 2021;272:120786.
96. Olson KE, Namminga KL, Schwab AD, et al. Neuroprotective activities of long-acting granulocyte-macrophage colony-stimulating factor (mPDM608) in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-intoxicated mice. Neurotherapeutics 2020;17:1861-77.
97. Kosloski LM, Kosmacek EA, Olson KE, Mosley RL, Gendelman HE. GM-CSF induces neuroprotective and anti-inflammatory responses in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine intoxicated mice. J Neuroimmunol 2013;265:1-10.
98. Hammond TR, Robinton D, Stevens B. Microglia and the brain: complementary partners in development and disease. Annu Rev Cell Dev Biol 2018;34:523-44.
99. McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988;38:1285-91.
100. Imamura K, Hishikawa N, Sawada M, Nagatsu T, Yoshida M, Hashizume Y. Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol 2003;106:518-26.
101. Bartels AL, Willemsen ATM, Doorduin J, de Vries EF, Dierckx RA, Leenders KL. [11C]-PK11195 PET: quantification of neuroinflammation and a monitor of anti-inflammatory treatment in Parkinson’s disease? Parkinsonism Relat Disord 2010;16:57-9.
102. Gerhard A, Pavese N, Hotton G, et al. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol Dis 2006;21:404-12.
103. Stokholm MG, Iranzo A, Østergaard K, et al. Assessment of neuroinflammation in patients with idiopathic rapid-eye-movement sleep behaviour disorder: a case-control study. Lancet Neurol 2017;16:789-96.
104. Członkowska A, Kohutnicka M, Kurkowska-Jastrzebska I, Członkowski A. Microglial reaction in MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) induced Parkinson’s disease mice model. Neurodegeneration 1996;5:137-43.
105. Hoenen C, Gustin A, Birck C, et al. Alpha-synuclein proteins promote pro-inflammatory cascades in microglia: stronger effects of the A53T mutant. PLoS One 2016;11:e0162717.
106. Hamza TH, Zabetian CP, Tenesa A, et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat Genet 2010;42:781-5.
107. Hendrickx DAE, van Eden CG, Schuurman KG, Hamann J, Huitinga I. Staining of HLA-DR, Iba1 and CD68 in human microglia reveals partially overlapping expression depending on cellular morphology and pathology. J Neuroimmunol 2017;309:12-22.
108. Beier EE, Neal M, Alam G, Edler M, Wu LJ, Richardson JR. Alternative microglial activation is associated with cessation of progressive dopamine neuron loss in mice systemically administered lipopolysaccharide. Neurobiol Dis 2017;108:115-27.
109. Milde S, van Tartwijk FW, Vilalta A, et al. Inflammatory neuronal loss in the substantia nigra induced by systemic lipopolysaccharide is prevented by knockout of the P2Y6 receptor in mice. J Neuroinflammation 2021;18:225.
110. Greenwood EK, Brown DR. Senescent microglia: the key to the ageing brain? Int J Mol Sci 2021;22:4402.
111. Streit WJ, Sammons NW, Kuhns AJ, Sparks DL. Dystrophic microglia in the aging human brain. Glia 2004;45:208-12.
112. Matsudaira T, Nakano S, Konishi Y, et al. Cellular senescence in white matter microglia is induced during ageing in mice and exacerbates the neuroinflammatory phenotype. Commun Biol 2023;6:665.
113. Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci 2007;8:57-69.
114. Kannarkat GT, Boss JM, Tansey MG. The role of innate and adaptive immunity in Parkinson’s disease. J Parkinsons Dis 2013;3:493-514.
115. Choi I, Zhang Y, Seegobin SP, et al. Microglia clear neuron-released α-synuclein via selective autophagy and prevent neurodegeneration. Nat Commun 2020;11:1386.
116. Guo M, Wang J, Zhao Y, et al. Microglial exosomes facilitate α-synuclein transmission in Parkinson’s disease. Brain 2020;143:1476-97.
117. Hong B, Ohtake Y, Itokazu T, Yamashita T. Glial senescence enhances α-synuclein pathology owing to its insufficient clearance caused by autophagy dysfunction. Cell Death Discov 2024;10:50.
118. Dermitzakis I, Theotokis P, Evangelidis P, et al. CNS border-associated macrophages: ontogeny and potential implication in disease. Curr Issues Mol Biol 2023;45:4285-300.
119. Schonhoff AM, Figge DA, Williams GP, et al. Border-associated macrophages mediate the neuroinflammatory response in an alpha-synuclein model of Parkinson disease. Nat Commun 2023;14:3754.
120. Utz SG, See P, Mildenberger W, et al. Early fate defines microglia and non-parenchymal brain macrophage development. Cell 2020;181:557-73.e18.
121. Silvin A, Uderhardt S, Piot C, et al. Dual ontogeny of disease-associated microglia and disease inflammatory macrophages in aging and neurodegeneration. Immunity 2022;55:1448-65.e6.
122. De Maeyer RPH, Chambers ES. The impact of ageing on monocytes and macrophages. Immunol Lett 2021;230:1-10.
123. Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A. Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat Rev Endocrinol 2018;14:576-90.
124. Qin XY, Zhang SP, Cao C, Loh YP, Cheng Y. Aberrations in peripheral inflammatory cytokine levels in Parkinson disease: a systematic review and meta-analysis. JAMA Neurol 2016;73:1316-24.
125. Williams-Gray CH, Wijeyekoon R, Yarnall AJ, et al. ICICLE-PD study group. Serum immune markers and disease progression in an incident Parkinson’s disease cohort (ICICLE-PD). Mov Disord 2016;31:995-1003.
126. Lindqvist D, Hall S, Surova Y, et al. Cerebrospinal fluid inflammatory markers in Parkinson’s disease - associations with depression, fatigue, and cognitive impairment. Brain Behav Immun 2013;33:183-9.
127. Lobanova E, Whiten D, Ruggeri FS, et al. Imaging protein aggregates in the serum and cerebrospinal fluid in Parkinson’s disease. Brain 2022;145:632-43.
128. Kouli A, Horne CB, Williams-Gray CH. Toll-like receptors and their therapeutic potential in Parkinson’s disease and α-synucleinopathies. Brain Behav Immun 2019;81:41-51.
129. Williams GP, Muskat K, Frazier A, et al. Unaltered T cell responses to common antigens in individuals with Parkinson’s disease. J Neurol Sci 2023;444:120510.
130. Chen H, Zhang SM, Hernán MA, et al. Nonsteroidal anti-inflammatory drugs and the risk of Parkinson disease. Arch Neurol 2003;60:1059-64.
131. Casper D, Yaparpalvi U, Rempel N, Werner P. Ibuprofen protects dopaminergic neurons against glutamate toxicity in vitro. Neurosci Lett 2000;289:201-4.
132. Hägg S, Jylhävä J. Sex differences in biological aging with a focus on human studies. Elife 2021;10:e63425.
133. Gillies GE, Pienaar IS, Vohra S, Qamhawi Z. Sex differences in Parkinson’s disease. Front Neuroendocrinol 2014;35:370-84.
134. Gullotta GS, Costantino G, Sortino MA, Spampinato SF. Microglia and the blood-brain barrier: an external player in acute and chronic neuroinflammatory conditions. Int J Mol Sci 2023;24:9144.
135. Wen W, Cheng J, Tang Y. Brain perivascular macrophages: current understanding and future prospects. Brain 2024;147:39-55.
136. Barcia C, Bautista V, Sánchez-Bahillo A, et al. Changes in vascularization in substantia nigra pars compacta of monkeys rendered parkinsonian. J Neural Transm 2005;112:1237-48.
137. Rite I, Machado A, Cano J, Venero JL. Blood-brain barrier disruption induces in vivo degeneration of nigral dopaminergic neurons. J Neurochem 2007;101:1567-82.
138. Elabi O, Gaceb A, Carlsson R, et al. Human α-synuclein overexpression in a mouse model of Parkinson’s disease leads to vascular pathology, blood brain barrier leakage and pericyte activation. Sci Rep 2021;11:1120.
139. Al-Bachari S, Naish JH, Parker GJM, Emsley HCA, Parkes LM. Blood-brain barrier leakage is increased in Parkinson’s disease. Front Physiol 2020;11:593026.
140. Kadry H, Noorani B, Cucullo L. A blood-brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 2020;17:69.
142. Olmedo-Díaz S, Estévez-Silva H, Orädd G, Af Bjerkén S, Marcellino D, Virel A. An altered blood-brain barrier contributes to brain iron accumulation and neuroinflammation in the 6-OHDA rat model of Parkinson’s disease. Neuroscience 2017;362:141-51.
143. Zhao C, Ling Z, Newman MB, Bhatia A, Carvey PM. TNF-α knockout and minocycline treatment attenuates blood-brain barrier leakage in MPTP-treated mice. Neurobiol Dis 2007;26:36-46.
144. Toledo JB, Arnold SE, Raible K, et al. Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer’s Coordinating Centre. Brain 2013;136:2697-706.
145. Malek N, Lawton MA, Swallow DM, et al. PRoBaND Clinical Consortium. Vascular disease and vascular risk factors in relation to motor features and cognition in early Parkinson’s disease. Mov Disord 2016;31:1518-26.
146. Verheggen ICM, de Jong JJA, van Boxtel MPJ, et al. Increase in blood-brain barrier leakage in healthy, older adults. Geroscience 2020;42:1183-93.
147. Montagne A, Barnes SR, Sweeney MD, et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015;85:296-302.
148. Bonte S, Vandemaele P, Verleden S, et al. Healthy brain ageing assessed with 18F-FDG PET and age-dependent recovery factors after partial volume effect correction. Eur J Nucl Med Mol Imaging 2017;44:838-49.
149. Daniel PM, Love ER, Pratt OE. The effect of age upon the influx of glucose into the brain. J Physiol 1978;274:141-8.
150. Mooradian AD, Morin AM, Cipp LJ, Haspel HC. Glucose transport is reduced in the blood-brain barrier of aged rats. Brain Res 1991;551:145-9.
151. Toornvliet R, van Berckel BNM, Luurtsema G, et al. Effect of age on functional P-glycoprotein in the blood-brain barrier measured by use of (R)-[11C]verapamil and positron emission tomography. Clin Pharmacol Ther 2006;79:540-8.
152. Ceafalan LC, Fertig TE, Gheorghe TC, et al. Age-related ultrastructural changes of the basement membrane in the mouse blood-brain barrier. J Cell Mol Med 2019;23:819-27.
153. Machin DR, Bloom SI, Campbell RA, et al. Advanced age results in a diminished endothelial glycocalyx. Am J Physiol Heart Circ Physiol 2018;315:H531-9.
154. Bell RD, Winkler EA, Sagare AP, et al. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 2010;68:409-27.
155. Stewart PA, Magliocco M, Hayakawa K, et al. A quantitative analysis of blood-brain barrier ultrastructure in the aging human. Microvasc Res 1987;33:270-82.
156. Banks WA, Reed MJ, Logsdon AF, Rhea EM, Erickson MA. Healthy aging and the blood-brain barrier. Nat Aging 2021;1:243-54.
157. Nikolakopoulou AM, Montagne A, Kisler K, et al. Pericyte loss leads to circulatory failure and pleiotrophin depletion causing neuron loss. Nat Neurosci 2019;22:1089-98.
159. Li Y, Xie L, Huang T, et al. Aging neurovascular unit and potential role of DNA damage and repair in combating vascular and neurodegenerative disorders. Front Neurosci 2019;13:778.
160. Allahyari RV, Clark KL, Shepard KA, Garcia ADR. Sonic hedgehog signaling is negatively regulated in reactive astrocytes after forebrain stab injury. Sci Rep 2019;9:565.
161. Propson NE, Roy ER, Litvinchuk A, Köhl J, Zheng H. Endothelial C3a receptor mediates vascular inflammation and blood-brain barrier permeability during aging. J Clin Invest 2021;131:140966.
162. Chen MB, Yang AC, Yousef H, et al. Brain endothelial cells are exquisite sensors of age-related circulatory cues. Cell Rep 2020;30:4418-32.e4.
163. Yousef H, Czupalla CJ, Lee D, et al. Aged blood impairs hippocampal neural precursor activity and activates microglia via brain endothelial cell VCAM1. Nat Med 2019;25:988-1000.
164. Gemechu JM, Bentivoglio M. T cell recruitment in the brain during normal aging. Front Cell Neurosci 2012;6:38.
165. Zhang X, Wang R, Chen H, et al. Aged microglia promote peripheral T cell infiltration by reprogramming the microenvironment of neurogenic niches. Immun Ageing 2022;19:34.
166. Berry K, Farias-Itao DS, Grinberg LT, et al. B and T lymphocyte densities remain stable with age in human cortex. ASN Neuro 2021;13:17590914211018117.
167. Yang AC, Stevens MY, Chen MB, et al. Physiological blood-brain transport is impaired with age by a shift in transcytosis. Nature 2020;583:425-30.
168. Pun PB, Lu J, Moochhala S. Involvement of ROS in BBB dysfunction. Free Radic Res 2009;43:348-64.
169. Ronaldson PT, Davis TP. Regulation of blood-brain barrier integrity by microglia in health and disease: a therapeutic opportunity. J Cereb Blood Flow Metab 2020;40:S6-24.
170. Bisht K, Okojie KA, Sharma K, et al. Capillary-associated microglia regulate vascular structure and function through PANX1-P2RY12 coupling in mice. Nat Commun 2021;12:5289.
171. Haruwaka K, Ikegami A, Tachibana Y, et al. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat Commun 2019;10:5816.
172. Huang W, Xia Q, Zheng F, et al. Microglia-mediated neurovascular unit dysfunction in Alzheimer’s disease. J Alzheimers Dis 2023;94:S335-54.
173. Ruan Z, Zhang D, Huang R, et al. Microglial activation damages dopaminergic neurons through MMP-2/-9-mediated increase of blood-brain barrier permeability in a Parkinson’s disease mouse model. Int J Mol Sci 2022;23:2793.
174. Lou N, Takano T, Pei Y, Xavier AL, Goldman SA, Nedergaard M. Purinergic receptor P2RY12-dependent microglial closure of the injured blood-brain barrier. Proc Natl Acad Sci U S A 2016;113:1074-9.
175. Zheng L, Guo Y, Zhai X, et al. Perivascular macrophages in the CNS: from health to neurovascular diseases. CNS Neurosci Ther 2022;28:1908-20.
176. Goldmann T, Wieghofer P, Jordão MJ, et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol 2016;17:797-805.
177. Lee D, Porras C, Spencer C, et al. Plasticity of human microglia and brain perivascular macrophages in aging and Alzheimer’s disease. medRxiv. [Preprint] Oct 26, 2023. [accessed on 2024 Mar 19]. Available from: https://www.medrxiv.org/content/10.1101/2023.10.25.23297558v1.
178. Drieu A, Du S, Storck SE, et al. Dominantly Inherited Alzheimer Network. Parenchymal border macrophages regulate the flow dynamics of the cerebrospinal fluid. Nature 2022;611:585-93.
179. Van Hove H, Martens L, Scheyltjens I, et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat Neurosci 2019;22:1021-35.
180. Huang X, Hussain B, Chang J. Peripheral inflammation and blood-brain barrier disruption: effects and mechanisms. CNS Neurosci Ther 2021;27:36-47.
181. Wang Y, Liu W, Geng P, et al. Role of crosstalk between glial cells and immune cells in blood-brain barrier damage and protection after acute ischemic stroke. Aging Dis 2023.
182. Galea I. The blood-brain barrier in systemic infection and inflammation. Cell Mol Immunol 2021;18:2489-501.
183. Wang X, Xue GX, Liu WC, et al. Melatonin alleviates lipopolysaccharide-compromised integrity of blood-brain barrier through activating AMP-activated protein kinase in old mice. Aging Cell 2017;16:414-21.
184. Gandhi K, Barzegar-Fallah A, Banstola A, Rizwan SB, Reynolds JNJ. Correction: Gandhi et al. Ultrasound-mediated blood-brain barrier disruption for drug delivery: a systematic review of protocols, efficacy, and safety outcomes from preclinical and clinical studies. Pharmaceutics 2022,14,833. Pharmaceutics 2023;15:5.
185. Tung YS, Vlachos F, Feshitan JA, Borden MA, Konofagou EE. The mechanism of interaction between focused ultrasound and microbubbles in blood-brain barrier opening in mice. J Acoust Soc Am 2011;130:3059-67.
186. Lipsman N, Meng Y, Bethune AJ, et al. Blood-brain barrier opening in Alzheimer’s disease using MR-guided focused ultrasound. Nat Commun 2018;9:2336.
187. Pineda-Pardo JA, Gasca-Salas C, Fernández-Rodríguez B, et al. Striatal blood-brain barrier opening in Parkinson’s disease dementia: a pilot exploratory study. Mov Disord 2022;37:2057-65.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Bendig J, Frank A, Reichmann H. Aging and Parkinson's disease: a complex interplay of vulnerable neurons, the immune system and the blood-brain barrier. Ageing Neur Dis 2024;4:5. http://dx.doi.org/10.20517/and.2023.36
AMA Style
Bendig J, Frank A, Reichmann H. Aging and Parkinson's disease: a complex interplay of vulnerable neurons, the immune system and the blood-brain barrier. Ageing and Neurodegenerative Diseases. 2024; 4(1): 5. http://dx.doi.org/10.20517/and.2023.36
Chicago/Turabian Style
Bendig, Jonas, Anika Frank, Heinz Reichmann. 2024. "Aging and Parkinson's disease: a complex interplay of vulnerable neurons, the immune system and the blood-brain barrier" Ageing and Neurodegenerative Diseases. 4, no.1: 5. http://dx.doi.org/10.20517/and.2023.36
ACS Style
Bendig, J.; Frank A.; Reichmann H. Aging and Parkinson's disease: a complex interplay of vulnerable neurons, the immune system and the blood-brain barrier. Ageing. Neur. Dis. 2024, 4, 5. http://dx.doi.org/10.20517/and.2023.36
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 6 clicks
Cite This Article 6 clicks

 Like This Article 5
likes
Like This Article 5
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.