
Genetically engineered pig models of neurological diseases

Abstract
Genetically modified animal models are commonly used for in vivo studies of human diseases. Mice are the most common animal models used in biomedical research, which have provided important insights into disease pathogenesis and are widely used to find treatments for diseases. However, due to the differences in the anatomical structure and physiological function between human and mouse brains, most genetically modified mouse models cannot fully recapitulate the overt and selective neuronal loss seen in age-dependent neurodegeneration diseases. While non-human primates (NHP) are closer to humans and have been used to model human disease, these models are difficult to be utilized at a large scale due to various limitations including their high costs, prolonged breeding time, community concerns for use of NHP, and high ethical standards. As an important animal resource in agriculture, pigs are also used as animal models in biomedical research. The central nervous system of pigs is highly similar to that of humans, making pig models suitable for investigating neurological diseases. The relatively short breeding period, large litter size, and established somatic cell transfer technology are advantages over NHP for using pigs to model human diseases. The recent development of gene editing tools allows one to more efficiently generate pig models that can precisely mimic genetic mutations in neurological diseases. In this review, we summarize recent advances in the use of pigs for modeling human neurological diseases, including new approaches for generating genetically modified pig models.
Keywords
INTRODUCTION
In vivo experiments using laboratory animals are essential for the verification of important findings from in vitro studies. In addition, animal models of human diseases are critical in revealing pathological changes and disease pathogenesis, which provide the theoretical basis for the development of treatments and therapeutic strategies. Small animal models such as mice and rats have been widely used in biomedical research, and animal models generated from mice have greatly advanced our understanding of the pathology and mechanisms of diseases. Small animals can partially mimic the symptoms and pathologic phenotypes of human disease, especially in extremely complex neurodegenerative diseases. That may be due to the considerable differences in development, aging, and fine structures between mouse and human brains. For example, the full development time for mouse brains is 21 days while primates’ brains need more than 150 days to reach full maturation[1]. The short lifespan of rodents is another major difference that may cause the different presentation of the neuropathology, since mice can only live for a little over two years, which is much shorter than the human’s average of 70 years. Therefore, the rapid development of the brain and the short lifespan of mice may cause neuronal cells to respond less strongly to the production of misfolded toxic proteins than do human neuronal cells. Differences in neural circuits and anatomical and physiological features between rodent and human brains suggest that we should explore other animal models to develop neurodegenerative diseases.
Undoubtedly, non-human primates (NHP) are ideal animal models that can closely mimic human diseases due to the high similarities between NHP and humans in genetics, physiology, development, social behaviors, and cognition. However, it is difficult to create a genetically modified NHP model when compared with small animals due to various factors, including long breeding cycles, lack of effective methods for genetic manipulations, high costs, community concerns, and high ethical standards. As a result, the first transgenic mouse model was generated as early as 1974[2], but the first genetically modified monkey model did not appear until 2001[3].
Considering the shortcomings of small animals and non-human primates in modeling human neurological diseases, pigs have some advantages over other species. Pig models have several unique features that make them a promising alternative animal model[4]. Pigs can produce larger litters and have a shorter maturation and reproduction time with fewer concerns about ethical issues and lower costs than non-human primates[5,6]. In regards to the similarity of pigs to humans, pigs are also highly close to humans in terms of anatomy, physiology, and metabolism[5]. As for the brain, the central nervous system of pigs is very similar to that of humans. For example, both human and pig brains have many sulci and gyri. Anatomically, the dorsal striata of the pig and human brains are both split into two distinct structures of the caudate nucleus and putamen, compared with a single structure in the rodent brain. In addition, the hippocampus in the pig brain more structurally resembles the human hippocampus than that in rodents. The timing of myelin formation in pig brains is also similar to that of humans during brain development[6] [Figure 1]. These similarities make the pig a better animal model for studying neurological diseases. In addition, pigs have the advantages of early sexual maturity (5-8 months), a short reproduction cycle between generations, and a larger litter size (about 10-12 piglets per litter)[7,8]. Moreover, fully established somatic cell nuclear transfer (SCNT) technology combined with recently developed genome editing technology has made it possible to efficiently generate genetically modified pig models[9] [Figure 2]. Here, we briefly discuss how to use related techniques to establish genetically modified pig models and review the established pig models for neurological diseases.
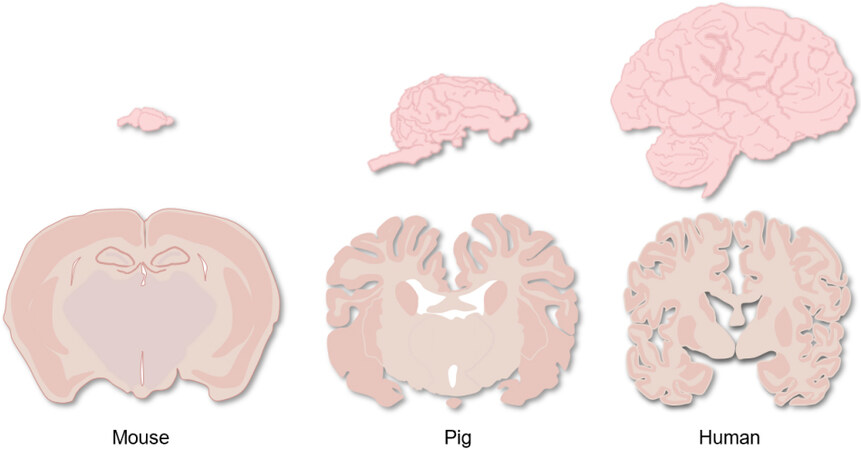
Figure 1. Comparison of brain structures of mouse, pig, and human.
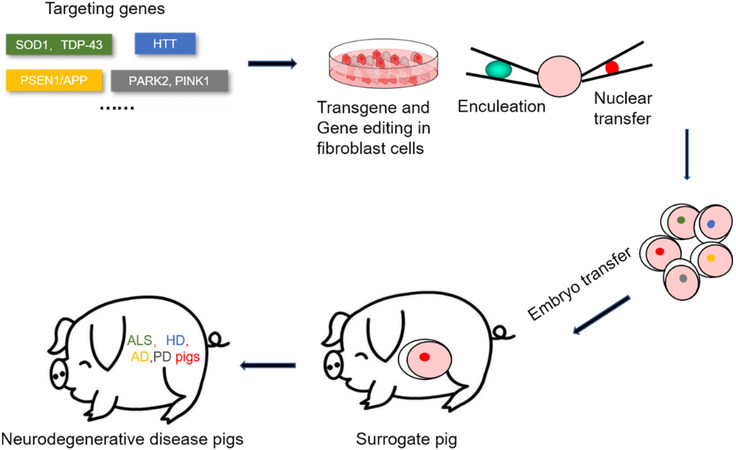
Figure 2. Flow chart of transgenic and gene editing using SCNT to construct neurodegenerative disease pig models. SCNT: Somatic cell nuclear transfer.
METHODS FOR GENERATING GENETICALLY MODIFIED PIG MODELS
For a long period of time, there have been two main methods to establish genetically modified pig models: embryonic microinjection and SCNT. Microinjection is a traditional method for creating transgenic animals and involves injecting DNA material directly into the pronucleus and transferring the early embryo into the surrogate mother to create a transgenic animal, which introduces transgenes randomly into the genome of the resulting offspring[10]. This method is fairly straightforward, but the efficiency of producing transgenic animals is relatively low, about 10% in mice, 4% in rabbits, and only 2%-3% in pigs[11,12]. Although several strategies have been used to improve the efficiency of embryonic microinjection, including pronuclei or cytoplasmic injection of DNA or mRNA[13,14], there are still many difficulties in using this method to generate genetically modified pig models. For example, due to the high lipid content and low transparency in pig oocytes[15], it is difficult to perform embryonic microinjection. In addition, this method will lead to random integration and poor precision of gene targeting. To improve the accuracy of gene editing, researchers developed a gene targeting strategy using homologous recombination (HR) in embryonic stem (ES) cells, which greatly improves the efficiency of generating gene-targeted animal models[16,17]. The lack of ES cells in pigs hinders the generation of precise genetically modified pig models. To overcome this difficulty, researchers firstly screen and identify the precisely targeted transgenes in cultured pig cells and then use them for SCNT, making it possible to establish gene-targeted pig models. However, the efficiency of HR in modifying pig somatic cells is very low, and the fatality rate is high due to the intrinsic genetic defects[18]. Later, an attempt to improve the efficiency of pig gene targeting was made by the application of several important technologies, including the delivery of gene-targeting vectors using recombinant adeno-associated virus (rAAV)[19,20].
GENOME EDITING TOOLS
Due to low targeting efficiency, for a long time, only a few transgenic pig models had been successfully established[21-24]. This situation was greatly improved with the development of new precise gene editing tools, which include zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein (Cas). ZFNs, composed of DNA-binding domains consisting of tandem zinc finger motifs with nuclease domains from the endonuclease FokI, can induce the targeted DNA double-stranded breaks (DSBs) that lead to DNA damage repair mechanisms[25,26]. Although ZFNs have been widely applied in many species, including plants, animals, and mammalian cells in culture[25], they have not been used to create large animal models.
TALENs are an alternative tool for genome engineering[27-29]. They are also fusion proteins of tandem repeats of a TAL effector protein and the FokI nuclease. TALENs induce the targeted DSBs that activate DNA damage response pathways and lead to gene knockout (KO) or knock-in (KI)[30]. As compared with ZFNs, TALENs are easier to design and synthesize, and some animal models of disease have been successfully established using TALENs[31].
Although ZFNs and TALENs have been applied to various species, CRISPR/Cas9 is now the most widely used genome editing tool for generating genetically modified animal models. The CRISPR/Cas9 system confers targeted gene editing by small RNAs that guide the Cas9 nuclease to the target site through base pairing[32]. When the complex is located at the targeting site of the genome, Cas9 cuts both strands at a precise location. Then, the repair mechanism kicks in to rejoin the damaged genomic DNA by non-homologous end joining (NHEJ) or homology-directed repair (HDR), which may result in mutations to inactivate or alter gene function. Based on this damage-repair mechanism, scientists have optimized the CRISPR/Cas9 system to create many genome editing models for small animals, such as mice[33], rats[34], and zebrafishes[35]. Large animal models such as pigs have also benefited from this technology. Here, we focus on genetically modified pig models of neurological diseases.
Pig models of amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is an adult-onset progressive neurodegenerative disease caused by the selective death of motor neurons (MNs). With the occurrence of aging, patients with ALS develop progressive loss of upper and lower MNs, muscle atrophy, and eventually paralysis, and they usually die within 3-5 years after the onset of symptoms[36,37]. Currently, the pathophysiological mechanism of ALS remains to be fully understood. Genetic studies have identified more than 30 gene mutations that are highly associated with the etiology of ALS, including copper/zinc superoxide dismutase 1 (SOD1) and TAR DNA-binding protein 43 (TDP-43). Mutations of these genes affect many cellular and molecular processes, leading to increased oxidative stress, mitochondrial dysfunction, excitatory toxicity, neuroinflammation, protein aggregation, and abnormal RNA metabolism. The neuropathology of ALS is characterized by protein aggregation and accumulation of ubiquitinated protein inclusion bodies in the neuronal cytoplasm. In most ALS patients, SOD1 and TDP-43 are the main components of these inclusion bodies, suggesting that SOD1 and TDP-43 are causative factors for the occurrence and development of ALS. Therefore, several studies have generated pig models that express mutant SOD1 or TDP-43 and showed ALS-like phenotypes.
Chieppa et al. produced an ALS pig model using SCNT in combination with transfected somatic cells expressing the G93A mutation of human SOD1[38]. In 2014, Yang et al. used similar techniques to generate transgenic pigs that express the same SOD1 mutation. The transgenic pigs developed age-dependent neuropathology and movement disorders, which recapitulate the features of the early disease symptoms seen in human ALS[9]. Moreover, transgenic mutant SOD1 pigs show intranuclear inclusions and an association of SOD1 with the nuclear protein PCBP1, which were not seen in mouse brains[9]. In addition to SOD1, researchers also established transgenic miniature pigs expressing mutant TDP-43. They found that transgenic TDP-43 was also distributed in the cytoplasm of neuronal cells resembling the pathology seen in human ALS brain tissues[39], which was not found in many transgenic TDP-43 mouse models[40-42]. Therefore, these pig models of ALS have a great value in studying the pathogenesis mediated by cytoplasmic mutant TDP-43 or intranuclear SOD1.
Pig models of Huntington’s disease
Huntington’s disease (HD) is an autosomal dominant and age-dependent neurological disorder characterized by motor dysfunction, cognitive decline, and psychological disturbance. Pathologically, HD is characterized by selective neurodegeneration, which preferentially occurs in the striatum. Most HD patients develop symptoms in middle age, and the symptoms worsen with age with patients usually dying 10-15 years after symptom onset[43]. HD results from a monogenetic mutation of a CAG repeat expansion in the exon1 of the gene Huntingtin (HTT). HTT is a multifaceted protein that is expressed ubiquitously and has numerous roles[44]. CAG repeat expansion (> 36 CAGs) in the HTT gene is translated to a polyglutamine (polyQ) expansion that causes HTT to misfold and aggregate in the brain. HD transgenic mice and HD-KI mice have been widely used, but their brains do not display the selective and striking neuronal loss seen in human HD patients[45].
In 2001, a transgenic pig model for HD was produced by pronuclear microinjection. However, the development of behavioral and neuropathological symptoms of HD in this transgenic pig model remains unclear[46,47]. In 2010, researchers used SCNT to successfully establish a transgenic HD pig model expressing N-terminal mutant HTT (1-208 amino acids) with 105Q. This pig model showed apoptosis in the brain and died postnatally. However, mice expressing the same transgene did not produce the brain pathology seen in pigs[48]. Later, another group used lentiviral transduction of pig embryos to establish a transgenic minipig model of HD expressing N-terminal mutant huntingtin (1-548 aa) under the control of human HTT promoter. However, this pig model did not develop motor deficits at up to 40 months of age, although mutant HTT mRNA and protein fragments were detected in the brain and peripheral tissues[49].
It is apparent that the phenotypes of transgenic HD pig models are dependent on the expression levels of transgenic N-terminal mutant HTT. It is important to create a pig model that expresses full-length mutant HTT at the endogenous level. With the development of CRISPR/Cas9 technology, precise gene editing of various animal species becomes possible[33], especially for the generation of large animal models[32]. To overcome the shortcomings of the transgenic pig model of HD, Yan et al. first used CRISPR/Cas9 to insert a large CAG repeat (150 CAGs) into the pig HTT locus in fibroblast cells, and then used SCNT to generate a HD knock-in pig model[50]. The brains of this pig model showed severe and preferential neurodegeneration in the medium spiny neurons in the striatum, an important pathological feature in HD patients. More importantly, the HD pig models displayed dance-like symptoms and breathing difficulties, which were similar to the symptoms in HD patients. Further, the pathogenic and neurologic features of HD pigs can be stably passed to offspring, enabling the establishment of a large animal model of HD for mechanistic study and drug screening.
Pig models of Alzheimer’s disease
The incidence rate of Alzheimer’s disease (AD) is increasing year by year with aging. Its early neurological symptoms are mainly memory loss and behavioral changes, and, in the late stage, the patients will have cognitive impairment, which severely affects daily life[51]. AD is usually divided into familial AD (FAD) and sporadic AD (SAD) according to different pathologies. Only about 5% of AD cases are FAD and are caused by mutations in β-amyloid precursor protein (APP), presenilin 1 (PS1), and/or presenilin 2 (PS2). Nearly 95% of patients with AD are classified as SAD, which is caused by a combination of genetic factors and environmental risk factors without documented familial history of AD[52]. The deposition of β-amyloid (Aβ) and hyperphosphorylation of Tau are the major pathological hallmarks, with other pathophysiologic changes including neuroinflammation, oxidative stress, and abnormal lipid metabolism. In addition to Aβ and Tau, apolipoprotein E4 (APOE4) and coulomb-receptor expressed on myeloid cells 2 (TREM2) are considered to be the risk factors[53]. Various mouse models of AD have been developed to mimic the symptoms of AD. However, due to the complexity of the neuropathology spectrum of AD, none of the available mouse models truly recapitulate the full spectrum of AD neuropathology, which includes Aβ deposition, synapse loss, inflammation, tau hyperphosphorylation, and neurofibrillary tangle formation[54]. To model the characteristics of AD in more human-like species, researchers injected Aβ oligomers into the lateral ventricle of macaques, which diffused into the brain and accumulated in several regions associated with memory and cognitive functions. They found that oligomer injections induced AD-like pathology with neurofibrillary tangle formation in the macaque brain, which was not found in small animal models[55]. Other researchers also used viral delivery of human 4R-tau to generate a tau-based rhesus monkey model of Alzheimer’s disease[56]. However, due to the long reproductive cycle of monkeys and immature cloning technology, it was difficult to obtain a large group of monkey models of AD through transgenic methods. Therefore, the establishment of transgenic pig models of AD is needed.
In 2009, Kragh et al. tried to develop a pig model of Alzheimer’s disease by expressing AD-causing dominant mutation APPsw. The transgene consisted of the cDNA of the neuronal variant of the human APP gene with the Swedish mutation. However, no disease phenotype was reported, although it was predicted that accumulation of the Aβ peptide in the brain might develop at the age of 1-2 years[57]. The same group also generated a transgenic miniature pig model expressing a cDNA of the AD-causing gene PSEN1M146I driven by an enhanced human UbiC promoter. However, no phenotypic data have been published yet[46,58]. To induce the neuropathology of the increased intraneuronal Aβ plaque formation, this group combined the mutation of PSEN1 and APP together to generate double transgenic Göttingen minipigs that carry one copy of a human PSEN1 cDNA with the Met146Ile (PSEN1M146I) mutation and three copies of a human AβPP695 cDNA with the Lys670Asn/Met671Leu (AβPPsw) double mutations. Their strategy successfully generated a pig model with an intraneuronal accumulation of Aβ42 in the brain between the age of 10 and 18 months, which may represent an early event in the pathogenesis of AD[59]. In 2017, another group used a retroviral multi-cistronic vector to generate an AD transgenic pig carrying three AD-related genes with a total of six well-characterized mutations: hAPP (K670N/M671L, I716V, and V717I), hTau (P301L), and hPS1 (M146V and L286P). They confirmed that transgenes were expressed at especially high levels in the brain. The levels of Aβ-40/42, total Tau, and GFAP were high in the brains of these transgenic animals as well. They proposed that more tests are needed in the future to find out if these pigs have age-dependent phenotypes of AD[60].
Pig models of Parkinson’s disease
Parkinson’s disease (PD), characterized by slowness of movement, limb stiffness, and tremors, is the second most common neurodegenerative disorder in the world. PD patients may also have issues such as cognitive issues, depression, anxiety, olfactory loss, and gastrointestinal disorder. The motor symptoms of PD are caused by the death of dopaminergic neurons in the substantia nigra[61]. Loss of dopamine neurons causes a drop in dopamine levels in the striatum, which leads to disrupted motor control[62]. Many mutations or variants in a number of genes, such as α-synuclein (SNCA), leucine-rich repeat kinase 2 (LRRK2), ten-induced kinase 1 (PINK1), arkin (PRKN), and protein deglycase (DJ-1), are found to increase the susceptibility to PD and have been used to create genetically modified animal models of PD[62,63]. However, many mouse models do not recapitulate the selective and progressive neurodegeneration seen in PD[64,65]. Although non-human primate models of PD have been established for investigation[66,67], it is difficult to establish a cohort of PD monkey models. Some teams thus explored the generation of pig models to study the neurological phenotypes of PD.
Yao et al. used TALENs combined with SCNT and embryo transfer to generate DJ-1 KO piglets by disrupting the PARK7 gene to model the phenotype of PD. Unfortunately, the piglets all died due to cloning defects, although DJ-1 protein was successfully repressed in all the detected tissues[68]. Another group used CRISPR/Cas9 combined with SCNT to generate PARK2 and PINK1 double-gene KO pigs. However, as with mouse PD models, no phenotypic symptoms of PD were observed in the seven-month-old live mutant pigs[69]. In 2016, Wang et al. generated a PD pig model using CRISPR/Cas9 system by simultaneously targeting three distinct genomic loci, Parkin/DJ-1/PINK1, in Bama miniature pigs. However, the piglets remained healthy with a normal growth rate, and no typical symptoms of Parkinson’s disease were observed in the 10-month-old live mutant pigs in this study[70].
BASE EDITING USED IN PIG MODELS
Although the CRISPR/Cas9 system has been widely used to facilitate genome editing, it could induce random insertions or deletions (indels) through error-prone NHEJ rather than the error-free HDR[35]. As a result, indels are obtained much more frequently at targeting sites than single-nucleotide substitutions. However, most human neurological diseases are induced by point mutations, rather than indels[71], which emphasizes the importance of the application of the genome-editing technique of base editing in the establishment of animal models of human neurological disease.
Base editing is a genome-editing technique that generates mutations at single-base resolution[72-74]. All four transition mutations, namely C to T, G to A, A to G, and T to C, can be inserted into the genome with the available CRISPR/Cas base editors (BEs). The cytosine base editor (CBE) can insert a C-G to T-A mutation, while the adenine base editor (ABE) can alter an A-T base pair into a G-C pair. In RNA, conversion of A to inosine (I) is also possible with the RNA base editor (RBE)[75].
The above advanced technologies have already been used to generate many genome editing models, especially in small animals and plants, such as mouse[76,77], rat[78], rabbit[79], sheep[80], rice[81], and wheat[82]. Some groups have also succeeded in applying this tool to large animals[83,84].
As for pigs, Li et al. first established pig models created via BE3, which separately targeted the TWIST2 gene and the TYR gene[85]. These pig models were able to reproduce the phenotypes of human diseases, which indicates that base editing systems provide a safer and more efficient approach to generating pig models that can precisely mimic point mutations of human diseases. Another study also indicated that using base editing technology was able to precisely introduce three gene (GGTA1, B4galNT2, and CMAH) base conversions into the pig genome with high efficiency[71]. In summary, there is enormous potential for establishing pig disease models of neurological disease through base editing because of its significant advantages compared with the traditional CRISPR/Cas9 system.
POTENTIAL LIMITATIONS OF USE OF PIG MODELS
Currently, pig models for neurodegenerative diseases provide considerable support for the analysis and treatment of such diseases in humans. In general, pig models have great potential to advance the study of human neurodegenerative diseases, from pathogenesis research to the development of drugs, and even as donors of tissues and organs.
In addition, while pig disease models have greatly accelerated advances in studying genetic diseases and testing drugs and treatments, there are still some problems. First, pigs require more space than rodents in animal facilities and, thus, higher maintenance costs. Second, due to their large size, surgical operations need to be performed by trained personnel, and because its brain is wrapped in a thick skull, the collection of brain tissue requires a high degree of proficiency of the operator, which increases the experimental cost to a certain extent. Third, because of their large size, behavioral tests will be more difficult. However, at present, various behavioral studies of pigs have been gradually improved, for instance, learning and memory study using novel object recognition tests; anxiety and depression measurement using open field[86]; neuropsychological screening for executive function, anxiety, willingness to explore a new environment, and locomotion using the open field test[87]; and motor ability measurement using a 3D kinematic gait analysis system[87].
CONCLUSION
A critical step in studying neurological diseases is to establish suitable animal models. Due to the complexity of neurological diseases, such as AD and PD, as well as the species differences between mice and humans, selective and overt neurodegeneration is not well modeled using mouse models. Pig models have great potential in modeling neurological diseases due to their close resemblance to the human nervous system, and several genetically modified pig models have been established for investigating neurodegenerative diseases [Table 1].
Examples of neurodegenerative disease pigs described in this article
| Pig models | Genes | Editing type | References |
| ALS pig | SOD1 | TG | [38] |
| ALS pig | SOD1 | TG | [9] |
| ALS pig | TDP-43 | TG | [39] |
| HD transgenic pig | HTT | TG | [47] |
| HD transgenic pig | N-mHTT(105Q) | TG | [48] |
| HD transgenic pig | HTT(1-548) | TG | [49] |
| HD KI pig | HTT | KI | [50] |
| AD transgenic pig | APPsw | PM | [57] |
| AD transgenic pig | PSEN1(M146I) | TG | [58] |
| AD transgenic pig | PSEN1, APP | PM | [59] |
| AD transgenic pig | hAPP, hTau, hPS1 | PM | [60] |
| PD pig | PARK7 | KO | [68] |
| PD pig | PARK2, PINK1 | M-KO | [69] |
| PD pig | Parkin, DJ-1, PINK1 | M-KO | [70] |
Pigs have very similar brain structure and function to humans. More importantly, pigs have sulci and gyri, and their brain volume is similar to that of humans, offering advantages over small animals for studying important brain diseases. Given their short reproductive cycle (5-6 months of sexual maturity) and multiple litter sizes (average of 7-8 piglets) as well as the availability of techniques for generating specific models of human diseases, pigs also have distinct advantages over non-human primates. Pigs can also be ethically used for translational research. For example, scientists and doctors recently successfully transplanted a pig heart into a patient with end-stage heart disease[88]. This work opened up a new avenue in the study of xenotransplantation.
The pig models can also be used for preclinical evaluation of stem cell therapy, gene therapy, and drug screening because their body size and metabolism are closer to humans than other species. Their relatively fast breeding and reproduction would provide a sufficient number of animals for evaluation of the therapeutic effects of drugs and other means. Considering the advanced gene editing tools available, we believe that genetically modified pig models will play a more important role in the studies of age-dependent neurological diseases in the future.
DECLARATIONS
AcknowledgementWe thank Dr. Xiao-Jiang Li for carefully editing the manuscript.
Authors’ contributionsWrote the review paper: Li C, Li J
Revised manuscript: Li S, Yan S
Conceived and designed experiments: Yan S, Lai L
All authors read and approved the final manuscript.
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by National Key Research and Development Program of China (2021YFA0805300), The National Natural Science Foundation of China (81922026, 82171244), and Guangzhou Key Research Program on Brain Science (202007030008, 202007030003).
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. Li XJ, Li S. Large animal models of Huntington’s disease. In: Nguyen HHP, Cenci MA, editors. Behavioral neurobiology of Huntington’s disease and Parkinson’s disease. Berlin: Springer Berlin Heidelberg; 2015. p. 149-60.
2. Jaenisch R, Mintz B. Simian virus 40 DNA sequences in DNA of healthy adult mice derived from preimplantation blastocysts injected with viral DNA. Proc Natl Acad Sci U S A 1974;71:1250-4.
3. Chan AW, Chong KY, Martinovich C, Simerly C, Schatten G. Transgenic monkeys produced by retroviral gene transfer into mature oocytes. Science 2001;291:309-12.
4. Prather R, Hawley R, Carter D, Lai L, Greenstein J. Transgenic swine for biomedicine and agriculture. Theriogenology 2003;59:115-23.
5. Roura E, Koopmans SJ, Lallès JP, et al. Critical review evaluating the pig as a model for human nutritional physiology. Nutr Res Rev 2016;29:60-90.
6. Kinder HA, Baker EW, West FD. The pig as a preclinical traumatic brain injury model: current models, functional outcome measures, and translational detection strategies. Neural Regen Res 2019;14:413-24.
7. Zou X, Ouyang H, Yu T, et al. Preparation of a new type 2 diabetic miniature pig model via the CRISPR/Cas9 system. Cell Death Dis 2019;10:823.
8. Zhao J, Lai L, Ji W, Zhou Q. Genome editing in large animals: current status and future prospects. Natl Sci Rev 2019;6:402-20.
9. Yang H, Wang G, Sun H, et al. Species-dependent neuropathology in transgenic SOD1 pigs. Cell Res 2014;24:464-81.
10. Gordon JW, Scangos GA, Plotkin DJ, Barbosa JA, Ruddle FH. Genetic transformation of mouse embryos by microinjection of purified DNA. Proc Natl Acad Sci U S A 1980;77:7380-4.
11. Hryhorowicz M, Lipiński D, Hryhorowicz S, Nowak-Terpiłowska A, Ryczek N, Zeyland J. Application of genetically engineered pigs in biomedical research. Genes (Basel) 2020;11:670.
12. Hammer RE, Pursel VG, Rexroad CE Jr, et al. Production of transgenic rabbits, sheep and pigs by microinjection. Nature 1985;315:680-3.
13. Chrenek P, Vasicek D, Makarevich AV, et al. Increased transgene integration efficiency upon microinjection of DNA into both pronuclei of rabbit embryos. Transgenic Res 2005;14:417-28.
14. Sumiyama K, Kawakami K, Yagita K. A simple and highly efficient transgenesis method in mice with the Tol2 transposon system and cytoplasmic microinjection. Genomics 2010;95:306-11.
15. Kikuchi K, Ekwall H, Tienthai P, et al. Morphological features of lipid droplet transition during porcine oocyte fertilisation and early embryonic development to blastocyst in vivo and in vitro. Zygote 2002;10:355-66.
16. Thomas KR, Capecchi MR. Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell 1987;51:503-12.
17. Mansour SL, Thomas KR, Capecchi MR. Disruption of the proto-oncogene int-2 in mouse embryo-derived stem cells: a general strategy for targeting mutations to non-selectable genes. Nature 1988;336:348-52.
18. Campbell KH. A background to nuclear transfer and its applications in agriculture and human therapeutic medicine. J Anat 2002;200:267-75.
19. Luo Y, Li J, Liu Y, et al. High efficiency of BRCA1 knockout using rAAV-mediated gene targeting: developing a pig model for breast cancer. Transgenic Res 2011;20:975-88.
20. Luo Y, Bolund L, Sørensen CB. Pig gene knockout by rAAV-mediated homologous recombination: comparison of BRCA1 gene knockout efficiency in Yucatan and Göttingen fibroblasts with slightly different target sequences. Transgenic Res 2012;21:671-6.
21. Dai Y, Vaught TD, Boone J, et al. Targeted disruption of the alpha1,3-galactosyltransferase gene in cloned pigs. Nat Biotechnol 2002;20:251-5.
22. Lai L, Kolber-Simonds D, Park KW, et al. Production of alpha-1,3-galactosyltransferase knockout pigs by nuclear transfer cloning. Science 2002;295:1089-92.
23. Rogers CS, Hao Y, Rokhlina T, et al. Production of CFTR-null and CFTR-DeltaF508 heterozygous pigs by adeno-associated virus-mediated gene targeting and somatic cell nuclear transfer. J Clin Invest 2008;118:1571-7.
24. Suzuki S, Iwamoto M, Saito Y, et al. Il2rg gene-targeted severe combined immunodeficiency pigs. Cell Stem Cell 2012;10:753-8.
26. Durai S, Mani M, Kandavelou K, Wu J, Porteus MH, Chandrasegaran S. Zinc finger nucleases: custom-designed molecular scissors for genome engineering of plant and mammalian cells. Nucleic Acids Res 2005;33:5978-90.
27. Christian M, Cermak T, Doyle EL, et al. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 2010;186:757-61.
28. Mussolino C, Cathomen T. TALE nucleases: tailored genome engineering made easy. Curr Opin Biotechnol 2012;23:644-50.
29. Joung JK, Sander JD. TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol 2013;14:49-55.
30. Yoshimi K, Mashimo T. Application of genome editing technologies in rats for human disease models. J Hum Genet 2018;63:115-23.
31. Liu H, Chen Y, Niu Y, et al. TALEN-mediated gene mutagenesis in rhesus and cynomolgus monkeys. Cell Stem Cell 2014;14:323-8.
32. Niu Y, Shen B, Cui Y, et al. Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell 2014;156:836-43.
33. Wang H, Yang H, Shivalila CS, et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 2013;153:910-8.
34. Ma Y, Zhang X, Shen B, et al. Generating rats with conditional alleles using CRISPR/Cas9. Cell Res 2014;24:122-5.
35. Kim H, Kim JS. A guide to genome engineering with programmable nucleases. Nat Rev Genet 2014;15:321-34.
36. Dion PA, Daoud H, Rouleau GA. Genetics of motor neuron disorders: new insights into pathogenic mechanisms. Nat Rev Genet 2009;10:769-82.
37. Boillée S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron 2006;52:39-59.
38. Chieppa MN, Perota A, Corona C, et al. Modeling amyotrophic lateral sclerosis in hSOD1 transgenic swine. Neurodegener Dis 2014;13:246-54.
39. Wang G, Yang H, Yan S, et al. Cytoplasmic mislocalization of RNA splicing factors and aberrant neuronal gene splicing in TDP-43 transgenic pig brain. Mol Neurodegener 2015;10:42.
40. Shan X, Chiang PM, Price DL, Wong PC. Altered distributions of Gemini of coiled bodies and mitochondria in motor neurons of TDP-43 transgenic mice. Proc Natl Acad Sci U S A 2010;107:16325-30.
41. Wils H, Kleinberger G, Janssens J, et al. TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A 2010;107:3858-63.
42. Xu YF, Gendron TF, Zhang YJ, et al. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J Neurosci 2010;30:10851-9.
43. Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP Jr. Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol 1985;44:559-77.
45. Wang CE, Tydlacka S, Orr AL, et al. Accumulation of N-terminal mutant huntingtin in mouse and monkey models implicated as a pathogenic mechanism in Huntington’s disease. Hum Mol Genet 2008;17:2738-51.
46. Fan N, Lai L. Genetically modified pig models for human diseases. J Genet Genomics 2013;40:67-73.
47. Uchida M, Shimatsu Y, Onoe K, et al. Production of transgenic miniature pigs by pronuclear microinjection. Transgenic Res 2001;10:577-82.
48. Yang D, Wang CE, Zhao B, et al. Expression of Huntington’s disease protein results in apoptotic neurons in the brains of cloned transgenic pigs. Hum Mol Genet 2010;19:3983-94.
49. Baxa M, Hruska-Plochan M, Juhas S, et al. A transgenic minipig model of Huntington’s Disease. J Huntingtons Dis 2013;2:47-68.
50. Yan S, Tu Z, Liu Z, et al. A Huntingtin knockin pig model recapitulates features of selective neurodegeneration in Huntington’s disease. Cell 2018;173:989-1002.e13.
51. Luo JE, Li YM. Turning the tide on Alzheimer’s disease: modulation of γ-secretase. Cell Biosci 2022;12:2.
52. Zhang L, Chen C, Mak MS, et al. Advance of sporadic Alzheimer’s disease animal models. Med Res Rev 2020;40:431-58.
53. Shi Y, Holtzman DM. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat Rev Immunol 2018;18:759-72.
54. Felice FG, Munoz DP. Opportunities and challenges in developing relevant animal models for Alzheimer’s disease. Ageing Res Rev 2016;26:112-4.
55. Forny-Germano L, Lyra e Silva NM, Batista AF, et al. Alzheimer’s disease-like pathology induced by amyloid-β oligomers in nonhuman primates. J Neurosci 2014;34:13629-43.
56. Beckman D, Chakrabarty P, Ott S, et al. A novel tau-based rhesus monkey model of Alzheimer’s pathogenesis. Alzheimers Dement 2021;17:933-45.
57. Kragh PM, Nielsen AL, Li J, et al. Hemizygous minipigs produced by random gene insertion and handmade cloning express the Alzheimer’s disease-causing dominant mutation APPsw. Transgenic Res 2009;18:545-58.
58. Jakobsen JE, Johansen MG, Schmidt M, et al. Generation of minipigs with targeted transgene insertion by recombinase-mediated cassette exchange (RMCE) and somatic cell nuclear transfer (SCNT). Transgenic Res 2013;22:709-23.
59. Jakobsen JE, Johansen MG, Schmidt M, et al. Expression of the Alzheimer’s disease mutations AβPP695sw and PSEN1M146I in double-transgenic göttingen minipigs. J Alzheimers Dis 2016;53:1617-30.
60. Lee SE, Hyun H, Park MR, et al. Production of transgenic pig as an Alzheimer’s disease model using a multi-cistronic vector system. PLoS One 2017;12:e0177933.
61. Raza C, Anjum R, Shakeel NUA. Parkinson’s disease: mechanisms, translational models and management strategies. Life Sci 2019;226:77-90.
62. Chia SJ, Tan EK, Chao YX. Historical perspective: models of Parkinson’s disease. Int J Mol Sci 2020;21:2464.
63. Grow DA, McCarrey JR, Navara CS. Advantages of nonhuman primates as preclinical models for evaluating stem cell-based therapies for Parkinson’s disease. Stem Cell Res 2016;17:352-66.
64. Kitada T, Pisani A, Porter DR, et al. Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc Natl Acad Sci U S A 2007;104:11441-6.
65. Dawson TM, Ko HS, Dawson VL. Genetic animal models of Parkinson’s disease. Neuron 2010;66:646-61.
66. Yang W, Liu Y, Tu Z, et al. CRISPR/Cas9-mediated PINK1 deletion leads to neurodegeneration in rhesus monkeys. Cell Res 2019;29:334-6.
67. Yang W, Guo X, Tu Z, et al. PINK1 kinase dysfunction triggers neurodegeneration in the primate brain without impacting mitochondrial homeostasis. Protein Cell 2022;13:26-46.
68. Yao J, Huang J, Hai T, et al. Efficient bi-allelic gene knockout and site-specific knock-in mediated by TALENs in pigs. Sci Rep 2014;4:6926.
69. Zhou X, Xin J, Fan N, et al. Generation of CRISPR/Cas9-mediated gene-targeted pigs via somatic cell nuclear transfer. Cell Mol Life Sci 2015;72:1175-84.
70. Wang X, Cao C, Huang J, et al. One-step generation of triple gene-targeted pigs using CRISPR/Cas9 system. Sci Rep 2016;6:20620.
71. Yuan H, Yu T, Wang L, et al. Efficient base editing by RNA-guided cytidine base editors (CBEs) in pigs. Cell Mol Life Sci 2020;77:719-33.
72. Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016;533:420-4.
73. Nishida K, Arazoe T, Yachie N, et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science 2016;353:aaf8729.
74. Gaudelli NM, Komor AC, Rees HA, et al. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017;551:464-71.
75. Molla KA, Yang Y. CRISPR/Cas-mediated base editing: technical considerations and practical applications. Trends Biotechnol 2019;37:1121-42.
76. Ryu SM, Koo T, Kim K, et al. Adenine base editing in mouse embryos and an adult mouse model of Duchenne muscular dystrophy. Nat Biotechnol 2018;36:536-9.
77. Lee HK, Willi M, Miller SM, et al. Targeting fidelity of adenine and cytosine base editors in mouse embryos. Nat Commun 2018;9:4804.
78. Ma Y, Yu L, Zhang X, et al. Highly efficient and precise base editing by engineered dCas9-guide tRNA adenosine deaminase in rats. Cell Discov 2018;4:39.
79. Liu Z, Chen M, Chen S, et al. Highly efficient RNA-guided base editing in rabbit. Nat Commun 2018;9:2717.
80. Zhang X, Li W, Liu C, et al. Alteration of sheep coat color pattern by disruption of ASIP gene via CRISPR Cas9. Sci Rep 2017;7:8149.
81. Hua K, Tao X, Yuan F, Wang D, Zhu JK. Precise A·T to G·C base editing in the rice genome. Mol Plant 2018;11:627-30.
82. Li C, Zong Y, Wang Y, et al. Expanded base editing in rice and wheat using a Cas9-adenosine deaminase fusion. Genome Biol 2018;19:59.
83. Li G, Zhou S, Li C, et al. Base pair editing in goat: nonsense codon introgression into FGF5 results in longer hair. FEBS J 2019;286:4675-92.
84. Zhou S, Cai B, He C, et al. Programmable base editing of the sheep genome revealed no genome-wide off-target mutations. Front Genet 2019;10:215.
85. Li Z, Duan X, An X, et al. Efficient RNA-guided base editing for disease modeling in pigs. Cell Discov 2018;4:64.
86. Haigh A, Chou JY, O’Driscoll K. Variations in the behavior of pigs during an open field and novel object test. Front Vet Sci 2020;7:607.
87. Netzley AH, Hunt RD, Franco-Arellano J, et al. Multimodal characterization of Yucatan minipig behavior and physiology through maturation. Sci Rep 2021;11:22688.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Li C, Li J, Lai L, Li S, Yan S. Genetically engineered pig models of neurological diseases. Ageing Neur Dis 2022;2:13. http://dx.doi.org/10.20517/and.2022.13
AMA Style
Li C, Li J, Lai L, Li S, Yan S. Genetically engineered pig models of neurological diseases. Ageing and Neurodegenerative Diseases. 2022; 2(3): 13. http://dx.doi.org/10.20517/and.2022.13
Chicago/Turabian Style
Li, Caijuan, Jun Li, Liangxue Lai, Shihua Li, Sen Yan. 2022. "Genetically engineered pig models of neurological diseases" Ageing and Neurodegenerative Diseases. 2, no.3: 13. http://dx.doi.org/10.20517/and.2022.13
ACS Style
Li, C.; Li J.; Lai L.; Li S.; Yan S. Genetically engineered pig models of neurological diseases. Ageing. Neur. Dis. 2022, 2, 13. http://dx.doi.org/10.20517/and.2022.13
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 61 clicks
Cite This Article 61 clicks

 Like This Article 20
likes
Like This Article 20
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.