
Ten selected articles in the field of neurodegenerative disease research
Our staff editors continue to share exciting, interesting, and thought-provoking reading material in the recommended articles series. This week, we would like to share 10 pick articles on neurodegenerative diseases.
Title: Parkinson’s disease patient-specific neuronal networks carrying the LRRK2 G2019S mutation unveil early functional alterations that predate neurodegeneration
Authors: G. Carola, D. Malagarriga, C. Calatayud, M. Pons-Espinal, L. Blasco-Agell, Y. Richaud-Patin, I. Fernandez-Carasa, V. Baruffi, S. Beltramone, E. Molina, P. Dell’Era, J. J. Toledo-Aral, E. Tolosa, A. R. Muotri, J. Garcia Ojalvo, J. Soriano, A. Raya, A. Consiglio
Type: Article from npj Parkinson's Disease
Abstract:
A deeper understanding of early disease mechanisms occurring in Parkinson’s disease (PD) is needed to reveal restorative targets. Here we report that human induced pluripotent stem cell (iPSC)-derived dopaminergic neurons (DAn) obtained from healthy individuals or patients harboring LRRK2 PD-causing mutation can create highly complex networks with evident signs of functional maturation over time. Compared to control neuronal networks, LRRK2 PD patients’ networks displayed an elevated bursting behavior, in the absence of neurodegeneration. By combining functional calcium imaging, biophysical modeling, and DAn-lineage tracing, we found a decrease in DAn neurite density that triggered overall functional alterations in PD neuronal networks. Our data implicate early dysfunction as a prime focus that may contribute to the initiation of downstream degenerative pathways preceding DAn loss in PD, highlighting a potential window of opportunity for pre-symptomatic assessment of chronic degenerative diseases.
Access this article: https://doi.org/10.1038/s41531-021-00198-3
Title: Identification of a novel interaction of FUS and syntaphilin may explain synaptic and mitochondrial abnormalities caused by ALS mutations
Authors: Shaakir Salam, Sara Tacconelli, Bradley N. Smith, Jacqueline C. Mitchell, Elizabeth Glennon, Nikolas Nikolaou, Corinne Houart, Caroline Vance
Type: Article from Scientific Reports
Abstract:
Aberrantly expressed fused in sarcoma (FUS) is a hallmark of FUS-related amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). Wildtype FUS localises to synapses and interacts with mitochondrial proteins while mutations have been shown to cause to pathological changes affecting mitochondria, synapses and the neuromuscular junction (NMJ). This indicates a crucial physiological role for FUS in regulating synaptic and mitochondrial function that is currently poorly understood. In this paper we provide evidence that mislocalised cytoplasmic FUS causes mitochondrial and synaptic changes and that FUS plays a vital role in maintaining neuronal health in vitro and in vivo. Overexpressing mutant FUS altered synaptic numbers and neuronal complexity in both primary neurons and zebrafish models. The degree to which FUS was mislocalised led to differences in the synaptic changes which was mirrored by changes in mitochondrial numbers and transport. Furthermore, we showed that FUS co-localises with the mitochondrial tethering protein Syntaphilin (SNPH), and that mutations in FUS affect this relationship. Finally, we demonstrated mutant FUS led to changes in global protein translation. This localisation between FUS and SNPH could explain the synaptic and mitochondrial defects observed leading to global protein translation defects. Importantly, our results support the ‘gain-of-function’ hypothesis for disease pathogenesis in FUS-related ALS.
Access this article: https://doi.org/10.1038/s41598-021-93189-6
Title: Drosophila tubulin polymerization promoting protein mutants reveal pathological correlates relevant to human Parkinson’s disease
Authors: Jing Xie, Shuting Chen, Jean C. Bopassa, Swati Banerjee
Type: Review from Scientific Reports
Abstract:
Parkinson’s disease (PD) is a progressive neurodegenerative disorder with no known cure. PD is characterized by locomotion deficits, nigrostriatal dopaminergic neuronal loss, mitochondrial dysfunctions and formation of α-Synuclein aggregates. A well-conserved and less understood family of Tubulin Polymerization Promoting Proteins (TPPP) is also implicated in PD and related disorders, where TPPP exists in pathological aggregates in neurons in patient brains. However, there are no in vivo studies on mammalian TPPP to understand the genetics and neuropathology linking TPPP aggregation or neurotoxicity to PD. Recently, we discovered the only Drosophila homolog of human TPPP named Ringmaker (Ringer). Here, we report that adult ringer mutants display progressive locomotor disabilities, reduced lifespan and neurodegeneration. Importantly, our findings reveal that Ringer is associated with mitochondria and ringer mutants have mitochondrial structural damage and dysfunctions. Adult ringer mutants also display progressive loss of dopaminergic neurons. Together, these phenotypes of ringer mutants recapitulate some of the salient features of human PD patients, thus allowing us to utilize ringer mutants as a fly model relevant to PD, and further explore its genetic and molecular underpinnings to gain insights into the role of human TPPP in PD.
Access this article: https://doi.org/10.1038/s41598-021-92738-3
Title: Age-dependent shift in the de novo proteome accompanies pathogenesis in an Alzheimer’s disease mouse model
Authors: Megan K. Elder, Hediye Erdjument-Bromage, Mauricio M. Oliveira, Maggie Mamcarz, Thomas A. Neubert, Eric Klann
Type: Article from Communications Biology
Abstract:
Alzheimer’s disease (AD) is an age-related neurodegenerative disorder associated with memory loss, but the AD-associated neuropathological changes begin years before memory impairments. Investigation of the early molecular abnormalities in AD might offer innovative opportunities to target memory impairment prior to onset. Decreased protein synthesis plays a fundamental role in AD, yet the consequences of this dysregulation for cellular function remain unknown. We hypothesize that alterations in the de novo proteome drive early metabolic alterations in the hippocampus that persist throughout AD progression. Using a combinatorial amino acid tagging approach to selectively label and enrich newly synthesized proteins, we found that the de novo proteome is disturbed in young APP/PS1 mice prior to symptom onset, affecting the synthesis of multiple components of the synaptic, lysosomal, and mitochondrial pathways. Furthermore, the synthesis of large clusters of ribosomal subunits were affected throughout development. Our data suggest that large-scale changes in protein synthesis could underlie cellular dysfunction in AD.
Access this article: https://doi.org/10.1038/s42003-021-02324-6
Title: SETBP1 accumulation induces P53 inhibition and genotoxic stress in neural progenitors underlying neurodegeneration in Schinzel-Giedion syndrome
Authors: Federica Banfi, Alicia Rubio, Mattia Zaghi, Luca Massimino, Giulia Fagnocchi, Edoardo Bellini, Mirko Luoni, Cinzia Cancellieri, Anna Bagliani, Chiara Di Resta, Camilla Maffezzini, Angelo Ianielli, Maurizio Ferrari, Rocco Piazza, Luca Mologni, Vania Broccoli, Alessandro Sessa
Type: Article from Nature Communications
Abstract:
The investigation of genetic forms of juvenile neurodegeneration could shed light on the causative mechanisms of neuronal loss. Schinzel-Giedion syndrome (SGS) is a fatal developmental syndrome caused by mutations in the SETBP1 gene, inducing the accumulation of its protein product. SGS features multi-organ involvement with severe intellectual and physical deficits due, at least in part, to early neurodegeneration. Here we introduce a human SGS model that displays disease-relevant phenotypes. We show that SGS neural progenitors exhibit aberrant proliferation, deregulation of oncogenes and suppressors, unresolved DNA damage, and resistance to apoptosis. Mechanistically, we demonstrate that high SETBP1 levels inhibit P53 function through the stabilization of SET, which in turn hinders P53 acetylation. We find that the inheritance of unresolved DNA damage in SGS neurons triggers the neurodegenerative process that can be alleviated either by PARP-1 inhibition or by NAD + supplementation. These results implicate that neuronal death in SGS originates from developmental alterations mainly in safeguarding cell identity and homeostasis.
Access this article: https://doi.org/10.1038/s41467-021-24391-3
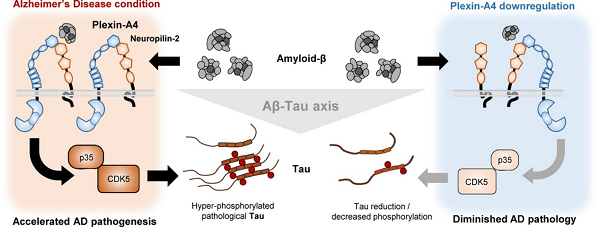
Title: Plexin-A4 mediates amyloid-β–induced tau pathology in Alzheimer’s disease animal model
Authors: Sunwoo Chung, Jinhee Yang, Haeng Jun Kim, Eun Mi Hwang, Wonik Lee, Kyujin Suh, Hayoung Choi, Inhee Mook-Jung
Type: Original Research Article from Progress in Neurobiology
Highlights:
• Plexin-A4 is a mediator of amyloid-β-induced tau pathology.
• We found a novel role of plexin-A4/neuropililn-2 complex as an amyloid-β receptor.
• Tau phosphorylation and gene expression is dependent on plexin-A4.
• Plexin-A4 downregulation protects Alzheimer’s disease model mice from pathological tau accumulation and memory impairment.
Abstract:
Amyloid-β (Aβ) and tau are major pathological hallmarks of Alzheimer’s disease (AD). Several studies have revealed that Aβ accelerates pathological tau transition and spreading during the disease progression, and that reducing tau can mitigate pathological features of AD. However, molecular links between Aβ and tau pathologies remain elusive. Here, we suggest a novel role for the plexin-A4 as an Aβ receptor that induces aggregated tau pathology. Plexin-A4, previously known as proteins involved in regulating axon guidance and synaptic plasticity, can bound to Aβ with co-receptor, neuropilin-2. Genetic downregulation of plexin-A4 in neurons was sufficient to prevent Aβ-induced activation of CDK5 and reduce tau hyperphosphorylation and aggregation, even in the presence of Aβ. In an AD mouse model that manifests both Aβ and tau pathologies, genetic downregulation of plexin-A4 in the hippocampus reduced tau pathology and ameliorated spatial memory impairment. Collectively, these results indicate that the plexin-A4 is capable of mediating Aβ-induced tau pathology in AD pathogenesis.
Graphical abstract:
Access this article: https://doi.org/10.1016/j.pneurobio.2021.102075
Title: Nilotinib restores memory function by preventing dopaminergic neuron degeneration in a mouse model of Alzheimer’s Disease
Authors: Livia La Barbera, Francescangelo Vedele, Annalisa Nobili, Paraskevi Krashia, Elena Spoleti, Emanuele Claudio Latagliata, Debora Cutuli, Emma Cauzzi, Ramona Marino, Maria Teresa Viscomi, Laura Petrosini, Stefano Puglisi- Allegra, Marcello Melone, Flavio Keller, Nicola Biagio Mercuri, Fiorenzo Conti, Marcello D’Amelio
Type: Original Research Article from Progress in Neurobiology
Highlights:
• Morphology and autophagic process are impaired in VTA DA neurons of Tg2576 mice.
• Tg2576 DA neurons show increased excitability and changes in neuronal conductances.
• Midbrain of Tg2576 mice shows increased activation of c-Abl tyrosine kinase.
• Nilotinib, a c-Abl inhibitor, ameliorates DA neuron loss and cell deficits.
• Nilotinib restores hippocampal DA levels and cognitive functions.
Abstract:
What happens precociously to the brain destined to develop Alzheimer’s Disease (AD) still remains to be elucidated and this is one reason why effective AD treatments are missing. Recent experimental and clinical studies indicate that the degeneration of the dopaminergic (DA) neurons in the Ventral Tegmental Area (VTA) could be one of the first events occurring in AD. However, the causes of the increased vulnerability of DA neurons in AD are missing.
Access this article: https://doi.org/10.1016/j.pneurobio.2021.102031
Title: Astrocytes and microglia in neurodegenerative diseases: Lessons from human in vitro models
Authors: Hannah Franklin, Benjamin E.Clarke, RickiePatani
Type: Review article from Progress in Neurobiology
Highlights:
• Astrocytes and microglia key fulfil homeostatic and immune functions in the CNS.
• Dysfunction of these cell types is implicated in neurodegenerative diseases.
• Understanding cellular autonomy and early pathogenic changes is a key goal.
• New human iPSC models will inform on disease mechanisms and therapy development.
Abstract:
Both astrocytes and microglia fulfil homeostatic and immune functions in the healthy CNS. Dysfunction of these cell types have been implicated in the pathomechanisms of several neurodegenerative diseases. Understanding the cellular autonomy and early pathological changes in these cell types may inform drug screening and therapy development. While animal models and post-mortem tissue have been invaluable in understanding disease processes, the advent of human in vitro models provides a unique insight into disease biology as a manipulable model system obtained directly from patients. Here, we discuss the different human in vitro models of astrocytes and microglia and outline the phenotypes that have been recapitulated in these systems.
Access this article: https://doi.org/10.1016/j.pneurobio.2020.101973
Title: Increased telomerase improves motor function and alpha-synuclein pathology in a transgenic mouse model of Parkinson’s disease associated with enhanced autophagy
Authors: Tengfei Wan, Emma J. Weir, Mary Johnson, Viktor I. Korolchuk, Gabriele C. Saretzki
Type: Original Research Article from Progress in Neurobiology
Highlights:
• Telomerase activators (TA) increase Tert expression in brains of a PD mouse model.
• Activator treatment improves PD motor symptoms: gait and balance.
• Activators reduce different forms of alpha-synuclein in brains of transgenic mice.
• Decreased autophagy markers LC3 and p62 suggest a better protein degradation.
• Our preclinical data suggest a use of TA to ameliorate PD-like symptoms.
Abstract:
Protective effects of the telomerase protein TERT have been shown in neurons and brain. We previously demonstrated that TERT protein can accumulate in mitochondria of Alzheimer’s disease (AD) brains and protect from pathological tau in primary mouse neurons. This prompted us to employ telomerase activators in order to boost telomerase expression in a mouse model of Parkinson’s disease (PD) overexpressing human wild type α-synuclein. Our aim was to test whether increased Tert expression levels were able to ameliorate PD symptoms and to activate protein degradation.
Access this article: https://doi.org/10.1016/j.pneurobio.2020.101953
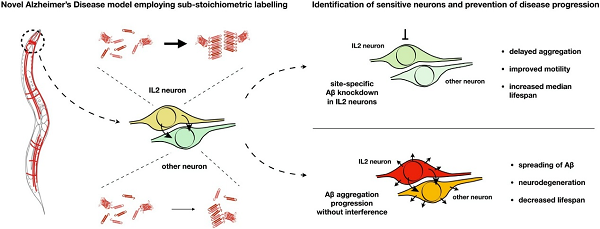
Title: Novel amyloid-beta pathology C. elegans model reveals distinct neurons as seeds of pathogenicity
Authors: Christian Gallrein, Manuel Iburg, Tim Michelberger, Alen Koçak, Dmytro Puchkov, Fan Liu, Sara Maria Ayala Mariscal, Tanmoyita Nayak, Gabriele S. Kaminski Schierle, Janine Kirstein
Type: Original Research Article from Progress in Neurobiology
Highlights:
• First in vivo animal model of sub-stoichiometric labelling of Aβ1−42 enables native amyloid fibril growth.
• Aβ1−42 spreads to distal cells and induces neurodegeneration in animal model.
• Aβ1−42 aggregation is heterogenic with a specific onset of aggregation pattern.
• Aggregation of Aβ1−42 starts in the cholinergic IL2 neurons of C. elegans.
• Targeted depletion of Aβ1−42 in IL2 neurons systemically ameliorates Aβ-pathology.
Abstract:
Protein misfolding and aggregation are hallmarks of neurodegenerative diseases such as Alzheimer’s disease (AD). In AD, the accumulation and aggregation of tau and the amyloid-beta peptide Aβ1−42 precedes the onset of AD symptoms. Modelling the aggregation of Aβ is technically very challenging in vivo due to its size of only 42 aa. Here, we employed sub-stoichiometric labelling of Aβ1−42 in C. elegans to enable tracking of the peptide in vivo, combined with the “native” aggregation of unlabeled Aβ1−42. Expression of Aβ1−42 leads to severe physiological defects, neuronal dysfunction and neurodegeneration. Moreover, we can demonstrate spreading of neuronal Aβ to other tissues. Fluorescence lifetime imaging microscopy enabled a quantification of the formation of amyloid fibrils with ageing and revealed a heterogenic yet specific pattern of aggregation. Notably, we found that Aβ aggregation starts in a subset of neurons of the anterior head ganglion, the six IL2 neurons. We further demonstrate that cell-specific, RNAi-mediated depletion of Aβ in these IL2 neurons systemically delays Aβ aggregation and pathology.
Graphical abstract:
Access this article: https://doi.org/10.1016/j.pneurobio.2020.101907




