
Tau-targeting therapy in Alzheimer's disease: critical advances and future opportunities


Abstract
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized by two pathological hallmark lesions: extracellular plaques composed of β-amyloid (Aβ) peptide and intracellular neurofibrillary tangles made up of highly phosphorylated tau protein. Over the past two decades, most disease-modifying therapies against AD have been developed mainly on the basis of the amyloid cascade hypothesis with a focus on Aβ. However, these agents yielded only limited benefits against disease progression, which prompts us to revitalize the long-neglected tau hypothesis. Tau protein is a microtubule-associated protein, which can stabilize microtubules, regulate microtubule assembly, and affect the morphology and growth of neuronal axons. Much more importantly, the degree of tau pathology is more closely related to cognitive decline in AD patients than that of Aβ pathology. Therefore, tau-targeting therapy seems to be a promising approach to combat AD. This review describes the research progress of tau-targeting therapy in AD, with an emphasis on immunotherapy. The current challenges and future perspectives in this field are also discussed.
Keywords
INTRODUCTION
Alzheimer’s disease (AD) is a progressive neurodegenerative disease characterized by progressive memory loss and cognitive impairment, accounting for 50%-75% of dementia cases. In the USA, an estimated 6.5 million people aged 65 and older had clinical AD in 2021, which will increase to 13.8 million by 2060 unless medical breakthroughs are developed to prevent, slow, or cure AD[1]. It is estimated that total medical expenditures for people suffering from AD and other dementias will increase from $321 billion in 2022 to just under $1 trillion by 2050[1]. In addition, patients with AD or other dementias have a higher chance of having more chronic diseases, which will generate more financial burden for treating comorbidities[2]; thus, it has become a major global health problem. The strategies for AD therapy mainly focus on two hallmark lesions: β-amyloid (Aβ), which forms extracellular plaque, and hyperphosphorylated tau, which forms intraneuronal neurofibrillary tangles (NFTs)[3].
It has long been widely accepted that changes in Aβ promote the disease process and drive tau pathology and neurodegeneration in AD[4]. Aβ can promote phosphorylation[5,6] or cleavage[7] of tau by activating related proteases, which in turn promotes tau aggregation and enhances tau-induced neurotoxicity. Beyond this triggering function, it has been commonly assumed that Aβ and tau act independently without specific interaction. However, accumulating evidence suggests there is a bidirectional interplay between Aβ and tau. Increased CSF tau levels were also demonstrated in APP/PS1 transgenic mice overexpressing Aβ, while tau deficiency ameliorated Aβ deposition[8]. The combination of Aβ and tau synergistically promotes the decrease of glucose metabolism, induces brain atrophy in AD brain, and leads to the declined cognitive function[9-11].
Currently, the drugs approved by the Food and Drug Administration for the treatments of AD include acetylcholinesterase inhibitors (donepezil, galantamine, and rivastigmine), NMDA receptor antagonist (memantine), and passive immunotherapy antibody aducanumab. However, these drugs only temporarily delay the disease progression of AD and ameliorate symptoms but cannot prevent or reverse the neuronal loss, brain atrophy, and the consequent progressive cognitive decline[12]. Since the introduction of the amyloid cascade hypothesis, most preventative or therapeutic candidates that were discovered and developed focus on Aβ. Aβ is mainly generated from amyloid precursor protein (APP) through sequential cleavage by β- and γ-secretase[13]. Therapeutic studies targeting Aβ have focused on inhibiting Aβ production by inhibiting β- or γ-secretase[14-16] and blocking Aβ aggregation[17], but these drugs yield only limited benefits and even induce serious adverse effects. Immunotherapy targeting Aβ was once a research hot spot; however, vaccine development has consistently struggled to bypass adverse outcomes such as excessive autoimmunity[18,19]. To date, most Aβ-targeting therapies have failed, which prompted us to re-examine the Aβ cascade hypothesis. Moreover, increasing lines of evidence suggest that tau pathology is more closely related to the cognitive decline in AD than Aβ, which revitalizes the long-neglected tau hypothesis, and more research attention has shifted from Aβ to pathological tau as a feasible target for disease intervention[20-23].
Tau protein is encoded by the microtubule-associated protein tau gene (MAPT) located on chromosome 17q21 and is abundant in neuronal axons. The adult human brain contains six tau isoforms ranging in size from 37 to 46 KD, which result from alternative splicing of exons 2, 3, and 10 of the MAPT gene [Figure 1]. Tau, an intrinsically disordered protein, can stabilize microtubules, regulate microtubule assembly, and affect the axonal morphology and growth of neurons[24]. Under pathological conditions, including AD, progressive supranuclear palsy (PSP), frontotemporal dementia (FTD), and other neurodegenerative diseases, tau’s abnormal assembly forms insoluble aggregates, accompanied by a series of neurodegenerative diseases such as synaptic dysfunction and nerve cell death[24,25]. As the course of AD progresses, abnormal tau aggregates to form NFTs existing in a characteristic distribution pattern, allowing the differentiation into six stages, namely Braak grades (transentorhinal stages I-II, clinically silent cases; limbic stage III-IV, incipient AD; and neocortical stages V-VI, fully developed AD)[26]. According to the pathological characteristics of tau, the prion-like aggregation and transmission of tau are proposed: the pathological tau takes itself as a template to induce the conformational changes of normal tau protein, so that it is easier to aggregate and induce more peripheral tau pathological changes, resulting in the spread of tau pathology to wider brain regions[27]. Studies have shown that, compared with the healthy control group, a large amount of tau protein is accumulated in the brain tissue of AD patients[28].

Figure 1. Schematic representation of the protein structures of tau. Six tau isoforms (2N4R, 2N3R, 1N4R, 1N3R, 0N4R, and 0N3R) of 352-441 aa are formed due to alternative splicing of exon 2 (E2), E3, and E10. Tau consists of four regions: N-terminus, proline-rich domain, microtubule-binding domain, and C-terminus. The expression of human tau is developmentally regulated: the 0N3R isoform is expressed only in the fetal brain, and all six isoforms are expressed in the adult brain. In the adult brain, the levels of the 3R and 4R forms are approximately equal, while the 0N, 1N, and 2N tau isoforms account for ~37%, ~54%, and ~9% of total tau, respectively.
Currently, tau-targeting therapy has become a hot topic in this field, including post-translational modifications (PTMs) of tau, inhibition of tau aggregation, stabilization of microtubules, and tau clearance by immunotherapy [Figure 2 and Table 1].
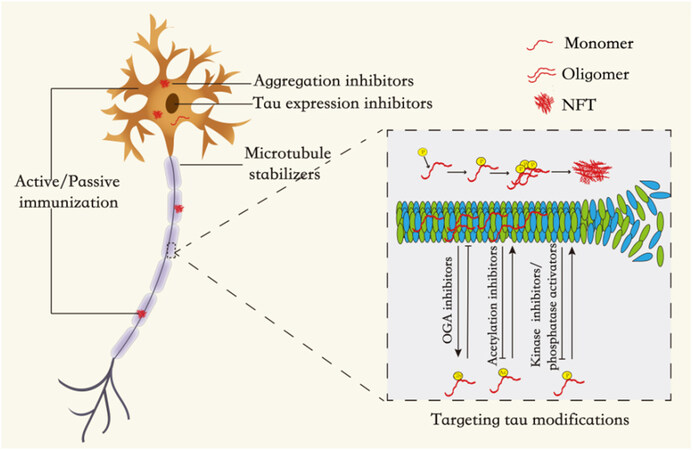
Figure 2. The therapeutic mechanism of targeting tau protein: the therapeutic research of targeting tau protein is mainly through targeting tau PTMs, the inhibition of tau protein aggregation and expression, stabilization of microtubules, and immunotherapy.
Research mechanisms and progress of related drugs targeting tau protein therapy
| Name | Synonyms | FDA status | Mechanism of action | Company |
| Targeting tau PTMs | ||||
| Tideglusib | NP031112, Nypta®, Zentylor™, GSK-3β inhibitor, NP12 | Alzheimer’s Disease (discontinued) | GSK-3β inhibitor | Zeltia Group |
| Lithium | MCI (phase 4), expected to be completed by 2023 | GSK-3β inhibitor | ||
| Sodium selenate | VEL015 | Alzheimer’s Disease (phase 2) | PP2A activators | |
| LY3372689 | Alzheimer’s Disease (phase 2) | O-GlcNAcase inhibitor | Eli Lilly & Co. | |
| Salsalate | Acetylation inhibitors | |||
| Aggregation inhibitors | ||||
| LMTM | TRx0237, LMT-X, Methylene Blue, TAI | Alzheimer’s Disease (phase 3) | Aggregation inhibitors | TauRx Therapeutics Ltd |
| ACI3024 | Tau MorphomerTM | Alzheimer’s Disease (inactive) | Aggregation inhibitors | AC Immune SA |
| Curcumin | Diferuloylmethane, Longvida™ | Alzheimer’s Disease (phase 2) | Aggregation inhibitors | Verdure Sciences |
| Tau expression inhibitors | ||||
| BIIB080 | IONIS-MAPTRx, ISIS 814907 | Alzheimer’s Disease (phase 1) | Inhibit the translation of tau mRNAs into protein | Biogen, IONIS Pharmaceuticals |
| Microtubule stabilizers | ||||
| Davunetide | NAP, AL-108 | MCI (discontinued) | Neuroprotective active agent that stabilizes microtubules in neurons | Allon Therapeutics Inc., Paladin Labs Inc. |
| Epothilone | BMS-241027 | Alzheimer’s Disease (discontinued) | Microtubule stabilizers | Bristol-Myers Squibb |
| Active immunization | ||||
| AADvac1 | Axon peptide 108 conjugated to KLH | Alzheimer’s Disease (phase2) | Targeting tau 294KDNIKHVPG GGS305 epitope | Axon Neuroscience SE |
| ACI-35 | Alzheimer’s Disease (phase2) | Targeting tau393VYKSPVVS GDTSPRHL408 epitope with the phosphorylation of Ser396/404 site | AC Immune SA, Janssen | |
| Passive immunization | ||||
| RG7345 | RO6926496 | Alzheimer’s Disease (discontinued) | A humanized monoclonal antibody targeting the tau phosphoepitope pS422 | |
| Gosuranemab | BIIB092, BMS-986168, IPN007 | Alzheimer’s Disease (discontinued) | A humanized IgG4 monoclonal antibody targeting the N-terminal region of tau protein | Biogen, Bristol-Myers Squibb |
| Semorinemab | RO7105705, MTAU9937A, RG6100 | Alzheimer’s Disease (phase2) | A monoclonal IgG4 antibody targeting extracellular tau, can bind the N-terminus of all six Tau isoforms of human tau | AC Immune SA, Genentech, Hoffmann-La Roche |
| Zagotenemab | LY3303560 | Alzheimer’s Disease (discontinued) | Recognizes the MC1 epitope, and binds soluble oligomers | Eli Lilly & Co. |
| JNJ63733657 | Mild AD (phase2), expected to be completed in March 2025 | A humanized IgG1 monoclonal antibody targeting MTBR of tau protein with a high affinity for pT217 | Janssen | |
| Bepranemab | UCB0107, UCB 0107, Antibody D | Alzheimer’s Disease (phase2), will run until November 2025. | A humanized IgG4 monoclonal antibody targeting tau235-250 near the MTBR of tau | Hoffmann-La Roche, UCB S.A. |
| PNT001 | Alzheimer’s Disease (phase 1) | A monoclonal antibody targeting the cis isomer of tau phosphorylated at Thr 231 | Pinteon Therapeutics | |
| BIIB076 | NI-105, 6C5huIgG1/l | Alzheimer’s Disease (phase 1) | A human recombinant IgG1 monoclonal antibody targeting mid-domain of tau | Biogen, Eisai Co., Ltd., Neurimmune |
| Tilavonema | C2N8E12, ABBV8E12, HJ9.3 | Alzheimer’s Disease (discontinued) | A humanized IgG4 antibody recognizes 25-30aa of tau and targets extracellular tau | AbbVie, C2N Diagnostics, LLC |
| E2814 | Alzheimer’s Disease (phase1/2), expected to be completed in April 2024 | A humanized IgG1 monoclonal antibody targeting the second and fourth HVPGG sequences of tau protein MTBR | Eisai Co., Ltd. | |
| Lu AF87908 | Alzheimer’s Disease (phase 1), expected to be completed in June 2022 | A humanized mouse IgG1 monoclonal antibody targeting tau386-408aa epitope with phosphorylated at Ser396 and Ser404 | Lundbeck |
TARGETING THE POST-TRANSLATIONAL MODIFICATIONS OF TAU
Pathological tau in AD has severe PTMs, and this process begins before the formation of NFTs, even decades before the onset of symptoms[29]. Not all PTMs are pathological, which is determined by the location, type, and amount of modification[30]. Hyperphosphorylation is the most studied PTM of tau and one of the earliest discovered modifications. Early studies found that aggregated tau isolated from AD brains had 3-4 times more overall phosphorylation level than healthy controls (2-3 mol per protein)[31]. Hyperphosphorylation of tau reduces the binding ability of tau to microtubules and reduces the stability of microtubules. The dissociated tau will self-aggregate, form oligomers and NFTs, disrupt the normal structure of cells, block intracellular material exchange, inhibit ubiquitin-proteasome activity, and cause aggregation, finally leading to neurodegeneration[32,33]. Both acetylation and N-glycosylation are also increased in AD. Acetylation inhibits the degradation of tau protein and promotes pathological tau aggregation and propagation[34-36], whereas N-glycosylation stimulates tau polymerization by promoting phosphorylation and conformational changes[37]. Nitration of the Tyr29 site of tau, found only in AD patients, significantly affects tau polymerization and facilitates tau aggregation[38]. The cleavage modification of caspase in AD patient brain also plays a key role. Caspases 2 and 3 cleave the C-terminus at Asp314 and Asp421, respectively, while caspase 6 cleaves the N-terminus[39,40]. In addition to the effect on microtubule binding and aggregation, C-terminal cleavage of tau is also associated with mitochondrial and synaptic damage[41,42]. However, not all PTMs accelerate tau pathology and AD progression. Ubiquitination promotes the degradation of tau by autophagy and proteasome, inhibits tau aggregation, and maintains microtubule stability[43]. Lysine methylation is part of the normal PTMs of tau in the human brain and can partially serve to protect against pathological tau aggregation[44]. Dityrosine (DiY) cross-links, another tau PTM induced by oxidative stress, was observed on human AD-derived tau oligomers and PHFs[45]. DiY cross-links of tau dGAE (tau297-391 fragment of the full-length tau) can facilitate the formation of non-toxic, soluble tau oligomers, inhibit the formation of β-sheets and further extension of prefibrils[46], and increase the insolubility and stability of tau fibrils in AD[45]. Different from the reported toxic tau oligomers, which are thought to be insoluble and β-sheet rich, tau oligomers formed by DiY cross-links are ThS-negative and random-coil rich[47]. These findings indicate that DiY cross-links seem to be beneficial in the progress of AD. Although tau has different isomers and the full-length tau has five tyrosines, the current research is mainly focused on the Y310 site of dGAE, and further research is needed to explore whether other tau isomers and tyrosine sites can form DiY cross-links and how they function.
Tau phosphorylation inhibitors
Tau protein has multiple phosphorylation sites, and its ability to bind to microtubules depends on the phosphorylation state. Hyperphosphorylated tau protein has less affinity for microtubules and even loses the ability to bind to microtubules, resulting in an unstable cytoskeleton. In addition, hyperphosphorylated tau can accumulate in neurons to form PHFs and eventually lead to neuronal death. Therefore, inhibition of tau hyperphosphorylation is crucial to maintaining normal neuronal physiological functions. The phosphorylation level of tau is mainly determined by the balance of various protein kinases and phosphatases, and the disruption of this balance will lead to the abnormal phosphorylation of tau observed in AD, especially the hyperactivation of glycogen synthase kinase 3β (GSK-3β) and cyclin-dependent kinase 5 (CDK5), as well as the inhibition of protein phosphatase-2A (PP2A)[48,49]. Inhibiting the activity of protein kinases and increasing the activity of phosphatases have been considered reasonable strategies for hampering tau pathology and AD therapy.
Protein phosphorylation inhibitors
GSK-3β inhibitors
Tideglusib (NP031112, Nypta®, Zentylor™, NP12) is an irreversible inhibitor of GSK-3β, a widely studied tau kinase, and its inhibition will counteract tau hyperphosphorylation[50]. Preclinical studies have shown that tideglusib can reduce a range of disease outcomes, including tau phosphorylation, Aβ deposition, neuron loss, and gliosis, in mouse entorhinal cortex and hippocampus and reverse spatial memory impairment in transgenic mice[51]. Moreover, the neuroprotective, anti-inflammatory, and neurogenesis-inducing effects of tideglusib have also been confirmed in animal models[52,53]. In a phase 2 trial involving mild to moderate AD patients, tideglusib had an acceptable safety profile for a short-term (26 weeks) treatment except for a transient increase in serum transaminase levels and diarrhea (14%-18% in active, 11% placebo) but failed to slow cognitive dysfunction, with only a small number of patients showing significant reductions of β-secretase in CSF[54,55]. Currently, the study of tideglusib in AD has been discontinued.
Lithium is a well-established drug for the treatment of bipolar disorder, and its role in AD has been studied due to its potential to inhibit GSK-3β[56,57]. On the one hand, after 10 weeks of lithium treatment in patients with mild or mild-to-moderate AD, clinical trials did not show significant improvement in cognitive scores and the therapeutic effects on GSK-3β activity or CSF-based biomarker concentrations[58]. On the other hand, in a 12-month double-blind trial in patients with amnestic mild cognitive impairment (MCI), the level of phosphorylated tau in CSF decreased in the lithium treatment group and cognitive ability was improved compared with the placebo control group. Moreover, the treatment of lithium was safe and well tolerated at a serum concentration range of 0.25-0.5 mmol/L[59,60]. In AD patients, a 15-month study of microdose lithium (300 μg/day) treatment also demonstrated its efficacy in preventing cognitive loss[61]. This is inconsistent with previous studies that failed to find a significant effect of lithium on cognition or AD-related biomarkers, possibly due to the high side effects[62] (renal and neurologic dysfunction, endocrine abnormalities, etc.) and withdrawal rate associated with higher lithium levels, shorter follow-up periods, and the included patients with cognitive degradation are related to the later stage. Taken together, the clinical data suggest that long-term microdose lithium therapy is necessary for a beneficial effect, and the use of microdose lithium may be able to resolve the toxicity problems that have pushed the development of the medication[62,63]. Currently, one phase 4 clinical trial of lithium in patients with MCI is underway and is expected to be completed by 2023[64].
AZD1080 is a novel and potent small molecule, orally bioavailable and brain-permeable selective GSK-3 inhibitor, which can inhibit tau phosphorylation in a dose-dependent manner in cells that overexpress tau protein and rescue synaptic plasticity deficits in rodent brain[65]. Meanwhile, AZD1080 has the ability to inhibit the GSK enzyme in humans, and the PK/PD profile demonstrates acceptable safety and tolerability in healthy volunteers after multiple oral ascending doses[65]. Injection of AZD1080 in MPTP mice suppressed the expression of phosphorylated tau and phosphorylated GSK-3β and improved motor functions[66]. Given the overall pharmacological profile, preclinical data, and the completion of additional toxicity studies, AZD1080 may have both disease-modifying and symptomatic potential in the treatment of AD and related tauopathies.
It is worth noting that, despite tau-phosphorylation, GSK-3β is also involved in various signaling pathways and physiological functions. Therefore, the non-specificity of GSK-3β inhibitors might contribute to the potential side effects as therapeutics against AD. Moreover, another important challenge and risk factor to overcome for a GSK-3β inhibitor to be converted into an effective and safe drug for AD treatment is its specific brain distribution.
CDK5 inhibitors
CDK5 is a proline-directed serine/threonine protein kinase activated by interacting with its activators, p35 and p39, or their cleavage products (p25 and p29, respectively), which governs various cellular processes in neurons. The dysregulation of CDK5 is largely due to the formation and accumulation of neurotoxic p25 and p29. The hyperactivation and mislocalization of CDK5 lead to hyperphosphorylation of its substrates[67]. Tau, as an important substrate of CDK5, can be phosphorylated by CDK5, then separates from microtubules and self-aggregates, and finally forms PHFs and insoluble NFTs in neurons[68]. Knockdown of CDK5 in triple transgenic (3xTg) AD mice, as well as inhibition of CDK5 with CDK5 inhibitory peptide in vitro, has been shown to reduce tau hyperphosphorylation and NFT formation[69]. Minocycline alleviates AD-like pathology and improves cognitive impairment by inhibiting the CDK5/p25 signaling pathway[70]. In the advanced stages of the disease, CDK5 primes phosphorylation sites on tau for GSK-3β and thereafter synergistically promote the GSK-3β-mediated tau hyperphosphorylation[71]. In contrast to this synergistic manner, one previous study reported that administration of CP681301, a CDK5 inhibitor, enhanced tau phosphorylation in p25-overexpressing transgenic mice. CDK5 can indirectly phosphorylate GSK-3β at S9 and inhibit its activity, suggesting that CDK5 inhibition may enhance tau phosphorylation through the activation of GSK3β[72,73]. Despite contradictory reports about the impacts of CDK5 on tau phosphorylation, another important concern is the tau-specificity of CDK5 inhibitors. Since CDK5 can phosphorylate molecules other than tau, more attention should be paid to developing tau-targeting agents with CDK5 inhibitory activity. In fact, to date, no successful small molecule candidates have been reported to selectively inhibit CDK5 kinase activity.
In addition to the kinases mentioned above, there are many other kinases that play an important role in the phosphorylation of tau. CAMP-dependent protein kinase A (PKA) is a serine/threonine protein kinase, and its activator forskolin can induce hyperphosphorylation of tau at Ser202/Thr205, Ser214, and Ser396[74]. However, PKA phosphorylates tau at Ser214, which reduces the pathological assembly of this protein[75]. The formation of PHFs is inhibited if tau is phosphorylated before GSK-3β, whereas PHFs are promoted if GSK-3β phosphorylates tau before PKA, suggesting that high PKA activity and low GSK-3β activity may inhibit the formation of NFTs[76]. AMP-activated protein kinase (AMPK) is a sensor of cellular stress and maintains energy homeostasis by controlling the activity of several metabolic enzymes. Studies have demonstrated that AMPK is dysregulated in the brains of AD patients, where it co-localizes with phosphorylated tau in pre-tangled and tangled neurons[77]. AMPK activation in mouse primary neurons increased tau phosphorylation at multiple sites, while AMPK inhibition rapidly reduced tau phosphorylation[77]. Phosphorylated AMPK accumulates in neuropil threads and dystrophic neurites around amyloid plaques and appears in over 90% of neurons with pre-tangles and tangles. In vitro experiments showed that AMPK could directly phosphorylate tau at Thr231 and Ser396/404, decreasing the microtubule binding affinity of tau[78]. However, one study also found that AMPK reduced tau phosphorylation, improved brain function, and inhibited GSK-3β activity in an AD-like model[79]. The microtubule-affinity regulating kinase 2 (MARK 2) is able to phosphorylate serine residues of tau protein’s KXGS motif[80]. Death-associated protein kinase (DAPK) activates MARK1 and MARK2, inhibits microtubule assembly, and destabilizes microtubules[81]. Furthermore, DAPK enhances tau toxicity and MARK-induced tau phosphorylation, and mice lacking DAPK show a reduction of tau phosphorylation in their brain[81].
Phosphatase activator
The phosphatases are mainly composed of PP2A, PP5, PP1, and PP2B, with proportions of 71%, 11%, 10%, and 7% of the total tau dephosphorylation activity, respectively, in the human brain[82]. Significant decreases in total phosphatase activity, PP2A activity, and PP5 activity toward tau were observed, but an increase in PP2B activity was also detected in AD brain[82]. The abnormal hyperphosphorylation of tau may be partly caused by the downregulation of PP2A activity, and its activity can be reduced by 50% in AD brain, which is consistent with the level of tau phosphorylation, whereas the activity of PP1 and PP5 is much less downregulated[82,83]. In vitro, co-incubation of tau aggregates with PP2A restored the binding of tau to microtubules to a level similar to the control group (about 80%)[84]. Therefore, PP2A is the major tau phosphatase, and its activators show promising potential for AD treatment.
Sodium selenate (VEL015): a preclinical study found that chronic treatment with low-dose VEL015 can reduce tau phosphorylation in cellular and mouse models of AD and prevent memory and motor deficits, NFTs formation, and neurodegeneration in tau transgenic mice[85]. In a 24-week, phase 2a, double-blind, randomized controlled trial in patients with mild to moderate AD, VEL015 was safe and well-tolerated[86]. However, no alterations of other AD biomarkers (CSF tau and Aβ levels) and cognitive performance were observed except for the reduction of neurodegeneration assessed by diffusion MRI[86]. In an open-label extension study, chronic VEL015 treatment (up to 23 months) in AD patients was also safe and well-tolerated. Moreover, the cognitive measures showed a slowed disease progression, but this was unable to be confirmed due to the uncontrolled trial design. Further investigations of VEL015 as a treatment for AD are warranted[84,87].
At present, few compounds have been proved to be effective in a tau transgenic mouse model, and fewer compounds have entered clinical trials. In addition to the general challenges of drug discovery and development for this type of disease (crossing the blood-brain barrier (BBB), target engagement, pharmacokinetics, long-term administration, etc.), developing kinase inhibitors with specificity for kinases (single-target vs. multi-target), formation of multiple substrates (off-target and potential side effects), or inhibition intensity (most tau kinases have many physiological functions) are also important issues to be addressed.
Targeting tau glycosylation
O-GlcNAcylation is a form of PTMs present in the nucleus and cytoplasm, in which a single N-acetylglucosamine (GlcNAc) is linked to the hydroxyl group of serine/threonine of proteins through O-glycosidic bonds. O-GlcNAc modification has important physiological significance, such as participating in signal transduction, gene expression, and cell cycle and proteasomal degradation. O-GlcNAc modification reduces the tendency of tau protein to form toxic aggregates[88,89]. O-GlcNAcase (OGA) catalyzes the removal of O-GlcNAc, whereas OGA inhibitors promote the O-GlcNAc of tau, prevent its aggregation, and stabilize tau in a soluble, nonpathogenic form. In different mutant human tau-expressing transgenic mice, the OGA inhibitor thiamet-G can increase O-GlcNAc-modification of tau and reduce the formation of NFTs and neuronal loss[90-92]. Impaired glucose uptake/metabolism can lead to AD and results in abnormal tau phosphorylation and NFTs formation through GSK-3β hyperactivation and reduction of O-GlcNAc[93]. Therefore, increasing the O-GlcNAc modification level has the potential to reduce tau phosphorylation, which helps to improve and slow down the progression of AD[94].
OGA inhibitor LY3372689, by increasing the level of O-GlcNAc modification, indirectly reduces the phosphorylation level of tau protein, prevents tau aggregation, and stabilizes tau in a soluble, nonpathogenic form, ultimately slowing the progression of AD[95]. In a phase 1 clinical trial, the drug was well-tolerated without serious adverse events or withdrawal due to adverse events. In September 2021, a phase 2 trial began to evaluate LY3372689 in 330 patients with early symptomatic AD who will receive a low or high dose of LY3372689 or a placebo for a period of 76-124 weeks. The trial is expected to be completed in June 2024.
Targeting tau acetylation
Salsalate is a small molecule nonsteroidal anti-inflammatory drug that inhibits acetylation of tau at Lys174 by the acetyltransferase p300. Acetylated tau is a blood biomarker of neurodegeneration induced by traumatic brain injury in mice and humans. Injury-induced neuronal tau acetylation leads to axonal damage and pathological tau mislocalization, while elevated NAD+ enhances the activity of the deacetylase Sirtl and blocks injury-induced tau acetylation, neurodegeneration, and neurobehavioral impairment[96]. Salsalate treatment for 2-3 months can prevent hippocampal atrophy, reduce tau pathology, and restore spatial memory deficits in PS19 mice[97]. In a phase 1 clinical trial of progressive supranuclear palsy (PSP), salsalate was safe and well tolerated, but it had no detectable effect on disease progression[98].
Targeting tau truncation
Enzyme-mediated (calpain, caspase, etc.) truncation of tau at the N-terminal (binding neuroplasmic membrane components) and C-terminal (binding axonal MT) is a contributing factor to AD pathology. Regardless of the protease, once tau is truncated, it is reasonable to presume an irreversible loss of its normal function. These fragments can promote tau aggregation, propagation between neurons through synapses, and expand nerve fiber degeneration to postsynaptic neurons, which has potential cytotoxicity[99,100]. Targeting the clearance of these fragments can significantly improve disease progression. Current therapeutic strategies for tau truncation are focused on targeting/blocking tau cleavage toxicity as well as clearance, including inhibitors of protease substrate interactions and antibody-mediated immune clearance. HJ8.5, a monoclonal antibody recognizing the N-terminal region of tau, is able to effectively in vitro block its seeding capacity and reduce pathological tau aggregates accumulation, microglial activation, and brain atrophy in six-month-old P301S tau transgenic mice, thereby improving their motor/sensory-motor and cognitive deficits[101,102]. 12A12, which selectively binds the pathologically relevant neurotoxic tau26-230 fragment of tau rather than reacting with its physiological form, is able to ameliorate the cognitive decline and memory impairment in Tg2576 and 3xTg mice[103]. Small molecule inhibitors targeting caspase-2 markedly blocked tau cleavage at the Asp314 site, prevented excessive accumulation of toxic tau fragments in dendritic spines, and restored excitatory neurotransmission in primary rat hippocampal neurons expressing the P301S tau variant[104]. At present, studies targeting tau fragment-mediated actions have only been performed in vitro, and further studies are needed to confirm whether it can be a potential therapeutic target for tau lesions.
Unlike other neurodegenerative diseases, tauopathies do not have potent genetic mutations of tau, and the pathological transformation from a highly soluble protein to an insoluble aggregate may be caused by extensive PTMs. PTMs are constantly and dynamically changing as the disease progresses. Compared with single PTM, combined PTMs seem to be more related to tau lesions, and they jointly affect the physicochemical properties of tau.
PREVENTING TAU AGGREGATION
The spread and accumulation of tau aggregates are related to neuronal loss and clinical symptoms in AD. The strategy of anti-protein aggregation therapy is to prevent the soluble tau protein from forming polymers and NFTs, so as to reduce the toxicity of tau protein and delay their dissemination. Therefore, targeting tau aggregation is a reasonable approach for AD treatment[105,106]. At the same time, the potential advantage of small molecule tau protein aggregation inhibitors that can target intracellular tau protein makes it the focus of research.
LMTM [Methylene Blue (MB), TRx0237, LMT-X, TAI]: MB showed an inhibitory effect on tau aggregation by blocking the tau-tau binding interaction through the repeat domain in vitro[107]. MB inhibits tau aggregation and rescues memory deficits in a mouse model of tauopathy[108]. Phase 2 clinical trials have demonstrated its cognitive benefits in patients with mild to moderate AD, but absorption of the highest dose is limited and tolerated poorly in the presence of starvation[109]. Moreover, MB lacked efficacy in a recent Phase 3 clinical trial[110]. Therefore, to overcome these limitations, methylmethionine (LMTM) - a reduced derivative of MB - was developed, which is more stable with better absorption, bioavailability, and tolerability[111]. In a phase 3 trial involving mild to moderate AD, LMTM failed to slow cognitive or functional decline, but after re-analysis of the data, the brain atrophy rate of patients with LMTM as monotherapy was significantly lower than that of mild AD. LMTM as monotherapy may have potential benefits, but due to factors such as pharmacokinetics, no difference was found between the clinical effects of high and low doses[110,112]. To confirm this hypothesis, a phase 3 trial of lower doses (8 and 16 mg daily) of LMTM is currently underway. The study will be completed in December 2022.
ACI-3024 (Tau MorphomerTM) is a small molecular inhibitor of tau aggregation that disrupts the β-sheet structure[113]. The mechanism is to prevent tau toxicity by reducing intracellular misfolding of tau protein and to slow tau protein spreading by preventing tangle formation or promoting tangle breakdown before tau protein tangles are secreted. Nanomolar concentrations of ACI-3024 alone cause a dose-dependent reduction in intracellular pathological tau and prevent the activation of microglia and neuronal death caused by PHFs. One-month treatment with ACI-3024 reduced aggregated and insoluble tau in the brains of tau lesion rTg4510 mice while decreased the activation of microglia and the total tau of CSF in proportion to the plasma drug concentration[114]. Oral treatment of ACI-3024 is being studied in neurodegenerative diseases and the phase 1 trial has been completed, but no preclinical information on ACI-3024 has been officially released.
Curcumin (diferuloylmethane, Longvida™) is the main ingredient of Indian turmeric spice, which is extracted from the rhizome of turmeric and has an excellent safety profile and a wide range of pharmacological activities with potential neuroprotective benefits, including anti-amyloid properties, inhibition of Aβ and tau aggregation, promotion of neurogenesis, and antioxidant and anti-inflammatory effects[115]. These effects suggest that curcumin may be useful in the treatment against tau-related neurodegeneration. However, curcumin has low bioavailability and can be rapidly degraded in vivo. Clinical trials in AD have shown no therapeutic effect of curcumin[116]. These results have led to the development of nanocarriers and analogs, and carrier devices such as liposomes and polymeric nanoparticles have shown promising potential[117]. J-147, an oral curcumin derivative, has been demonstrated to easily cross BBB and enhance memory in aged AD mice[118]. In addition, purpurin[119], ginseng[120], and many other natural compounds are also being investigated, which will offer great potential for the treatment of AD.
In addition to inhibiting tau aggregation, there are still some inhibitors reported to act as dual inhibitors of Aβ and tau. Curcumin can effectively disaggregate Aβ as well as preventing fibril and oligomer formation at low concentration[121]. At the same time, some small molecules (e.g., thiophene and ortho catechol) that have not yet entered clinical research are also playing the same role[122,123]. Many single-target drugs for Aβ and tau have failed in different stages of drug development. Therefore, multi-functional drug molecules targeting multiple targets are necessary for the treatment of AD.
However, choosing the appropriate inhibitor of pathological protein aggregation is a big challenge. It may be detrimental if the inhibitors dissociate large aggregates into smaller (toxic) oligomers or inhibit the inability of toxic oligomers to continue to form fewer toxic fibrils.
REDUCING TAU EXPRESSION
BIIB080 (IONIS-MAPTRx, ISIS 814907): tau-targeted antisense oligonucleotide (ASO) therapy has been shown to inhibit the translation of tau mRNAs and ameliorate toxin-induced seizures, neuronal loss, and neurofibrosis in adult tau-transgenic mouse models[124]. Infusion of BIIB080 into the CSF of cynomolgus monkeys reduced tau mRNA across different brain regions, and CSF tau levels following ASO exposure were correlated with hippocampal tau levels[124,125]. The results of one phase 1 clinical trial show that BIIB080 could reduce the total tau and pTau-181 protein levels in CSF of mild AD patients in a dose-dependent manner, with good safety and tolerability. Phase 2 trials will be conducted in mid-2022. Although BIIB080 can non-selectively reduce the levels of both normal and pathological tau protein, there are still some safety concerns. Studies showed that tau knockdown can impair the repulsive response of the growth cone[126], delay neuronal maturation[127], accelerate neuronal branching[128], and hyperpolarize neuronal membrane potential[129]. In addition, the reduction of tau may decrease mitochondrial mobility, increase the number of abnormal mitochondria[130], and lead to a loss of its nucleic acid safeguarding functions and increase in DNA and RNA oxidative damage[131]. However, only mild phenotypic changes (such as muscle weakness and hyperactivity) and delayed neuronal differentiation were shown in the tau knockout mouse model[127,132]. The reason may be that the changes caused by tau deletion during neuronal development may be compensated by the increased expression of other microtubule-associated proteins. Hence, tau pathologies are primarily caused by gain-of-function abnormalities caused by tau misregulation. We need to consider its impact on normal tau physiological function when targeting tau protein reduction.
PROMOTING MICROTUBULE STABILIZATION
Microtubules are key cytoskeletal elements in living cells, which are essential for axonal transport, synaptic transmission, and maintenance of neuronal morphology. Tau protein normally stabilizes microtubules, but hyperphosphorylation causes loss of its function. Microtubule dysfunction and subsequent axonal transport disorders in neurons are believed to play a prominent role in the neurodegeneration of AD[133,134].
Davunetide (NAP, AL-108) is derived from a growth factor called activity-dependent neurotrophic protein, which has highly effective neuroprotective activity. It is a peptide that can modulate the pool of microtubules in neurons and glial cells, although its exact mechanism of action is unclear[135,136]. Davunetide is related to the memory and cognitive ability of mice. In the 3xTg mouse model of AD, davunetide reduces the amyloid accumulation and tau hyperphosphorylation and improves the behavioral performance in the Morris water maze[137,138]. Davunetide entered the clinical trial after a successful preclinical study showing positive results[136,139]. The phase 2 trial of intranasal administration of davunetide in prodromal AD subjects showed good safety, but there was no significant improvement in cognitive score[140]. At present, the experiment has been terminated.
Epothilone D (BMS-241027) is a small molecule microtubule stabilizer isolated from myxobacterium Sorangium cellosum for the treatment of AD. Epothilone D can cross the BBB and reverse behavioral and cognitive deficits, clear tau pathology, and curb neuron loss in P301S tau transgenic mice[141,142]. A phase 1 trial of weekly infusion of epothilone D (0.003, 0.01, and 0.03 mg/kg) in patients with mild AD for nine weeks was conducted to evaluate the tolerability and pharmacology of BMS-241027. The clinical trial also monitored adverse effects and measured whether drugs changed the N-terminal fragment concentration of tau in CSF and cognitive performance. However, the study was terminated without data release.
TARGETING TAU INTERCELLULAR TRAFFICKING
It has been found that the intercellular transmission of tau also plays an important role in the development of AD. Therefore, research targeting tau transmission mechanisms seems to offer opportunities for AD treatment and drug development.
In AD, pathological neurofibrillary tau aggregates display an accumulative pattern that begins in the entorhinal cortex and travels through connected pathways to cortical areas, with cognitive impairment manifesting itself when tau inclusions reach the hippocampus. The presence of tau in the CSF of AD patients has long been thought to be simply the result of the passive release of degenerated and dead neurons. Previous studies found that injection of mutant P301S tau-expressing mice’s brain extracts into the brains of transgenic wild-type tau-expressing mice induced the assembly of wild-type human tau into filaments and the spread of pathology from the injection site to adjacent brain regions[143]. In addition, young PS19 tau transgenic mice injected with pathological tau promote tau lesions in a time- and dose-dependent manner[144]. Growing evidence suggests that, in most neurodegenerative diseases, misfolded pathological tau proteins act as seeds (oligomers and protofibrils), which then recruit and convert normal monomeric tau proteins within the cytoplasm of recipient neurons into misfolded pathological tau proteins. The misfolded and pathological tau proteins then travel along anatomically related neural pathways in the brain to adjacent normal cells or synaptically interconnected cells, spreading pathology from affected cells to healthy cells and to previously unaffected brain regions, promoting disease progression[145-148]. Current research speculates that the main mechanisms of cell-to-cell transmission of misfolded pathological proteins mainly include the following cellular processes: (1) Donor neurons can release tau seeds by exocytosis or vesicles such as exosomes that fuse with and deliver their contents to recipient neurons. Extracellular tau seeds can be internally taken up by recipient neurons through endocytosis; (2) In addition to the intercellular transfer of tau via the extracellular space, direct intercellular mechanisms via tunneling nanotubes allow for the transfer of cytoplasmic tau directly from the cytoplasm of one cell to another. Tau, which is simultaneously recruited and aggregated on the cellular leaflets of plasma membrane (PM), interacts directly with specific lipids in cholesterol/sphingomyelin/PI(4,5)P2-rich membrane microdomains, and then penetrates through the PM and releases from the PM facilitated by cell surface heparan sulfate proteoglycans (HSPGs).
Currently, identifying the mechanisms of tau propagation has led to the development of monoclonal antibodies or other compounds that specifically bind to, sequester, or disassemble tau conformations relevant to pathological propagation and may delay disease progression in tauopathies. In addition, inhibition of the process by which tau interacts with PM lipids or cell surface HSPGs may reduce the extracellular tau internalization. However, there are many unresolved issues and challenges regarding the mechanisms of tau propagation. Tau has a variety of pathological conformations, but it is unclear whether specific secretory pathways are preferred by certain types of tau aggregates. Moreover, the receptor proteins involved in the interactions between tau and cell membrane are largely unknown. The mechanism of how normal tau proteins are transformed into misfolded tau aggregates by using pathological tau seeds as templates is also unclear and requires urgent and extensive research to confirm.
PROMOTING TAU CLEARANCE
Autophagic clearance of tau aggregation
The ubiquitin-proteasome system and the autophagy-lysosome pathway (ALP) are the two main modes of tau degradation, while abnormally phosphorylated tau and PHF tau are mainly degraded by ALP[43]. In many neurodegenerative diseases, autophagy dysfunction disrupts the efficient clearance of misfolded proteins and cytoplasmic oligomers. A complete autophagic process consists of three consecutive steps, namely induction, autophagosome formation, autophagosome-lysosome fusion, and degradation[149]. The regulation of autophagy involves complex signaling pathways that can be divided into two main aspects: an mTOR-dependent approach and an mTOR-independent approach; however, both regulatory pathways have been found to be aberrant in AD[150].
Inhibition of MTORC1 and autophagic activity directly correlate with tau clearance; activation of autophagy with specific 12/15-lipoxygenase inhibitors or drugs such as rapamycin significantly inhibits tau aggregation and reduces insoluble and phosphorylated tau protein levels in the brain of AD mice, reducing tau lesions and cognitive dysfunction in neuronal cells[151]. Autophagy-based mTOR inhibitors (OSI-027, AZD2014, and AZD8055) potently downregulate tau phosphorylation, prevent the formation of insoluble tau, and promote autophagy-mediated tau clearance, thereby reducing tau-mediated neuronal stress vulnerability[152].
Selenium-methionine activates autophagy via the AMPK-mTOR pathway and then promotes tau clearance from neurons to improve cognitive performance in AD model mice[153]. Nuclear factor E2-related factor2 promotes autophagy and autolysosomal clearance through directly regulating the expression of Bcl-2 associated athanogene 3, autophagic bridging proteins NBR1 and NDP52, and autophagy protein p62, so as to play important regulatory roles in autophagy-mediated degradation of phosphorylated tau protein[154].
miR-132/212 targets tau mRNA to regulate tau expression, whereas deletion of miR-132/212 induces tau aggregation in mice expressing endogenous or human mutant tau. Moreover, treatment of AD mice with miR-132 mimics partially restores memory function and tau metabolism. miR-132/212 levels are correlated with insoluble tau and cognitive impairment in humans[155]. Blocking cholesterol acyltransferase expression with inhibitors revealed enhanced autophagy and induced autophagosome formation in AD mouse models, accompanied by a decrease in phosphorylated tau[156].
Therefore, all these research findings support that reducing the accumulation of intracellular aggregate proteins through autophagic upregulation could be a promising therapeutic strategy for most neurodegenerative diseases. However, low efficacy and severe side effects due to the lack of selectivity towards diseased cells and potential autophagic cell death have limited the further development of autophagic modulators. A recent tauopathy-homing nanocomponent with autophagy-activating capacity appears to address part of the problem by binding to hyperphosphorylated and/or aggregated tau and selectively aggregating in cells with undergoing tauopathy, further specifically promoting the clearance of pathogenic tau by stimulating autophagic flux and thus rescuing neuronal viability and cognitive function in AD rats[157]. Although activation of autophagy is a promising therapeutic approach in neurodegenerative diseases with impaired lysosomal clearance, overactivation of autophagy may contribute to disease progression. There is a dual role for autophagy in neurodegenerative diseases: in the early stages of AD, autophagy is increased, helping the removal of abnormally folded proteins and preventing further development of AD; however, in the advanced stages of the disease, the autophagic system is abnormal and the clearance of autophagosomes does not keep up with the formation of autophagosomes, leading to the formation and aggregation of neurotoxic Aβ and tau protein oligomers[150,158]. Although various autophagy enhancers have been identified to slow the progression of AD and improve cognitive performance, the beneficial effects in AD patients are still limited. Deep thoughts and a comprehensive understanding of the role of autophagy in the pathogenesis of AD are still required to provide new theoretical and even therapeutic targets for clinical trials of anti-AD drugs.
Active immunotherapy against tau pathology
Earlier tau-targeting therapies primarily focused on inhibiting tau phosphorylation and aggregation or stabilizing microtubules. However, these approaches have mostly failed due to toxicity and/or lack of efficacy. Most of the current therapies targeting tau are moving to immunotherapy. Both active and passive immunization are designed to form antibodies targeting the pathological conformation of tau without responding to non-pathological tau. Promoting the clearance of abnormal tau is expected to reduce neuronal loss and ameliorate clinical symptoms[159,160]. The first vaccine for tau active immunotherapy was designed against recombinant tau protein, which was inoculated into C57BL/6 wild-type mice. Although it can induce tau antibodies, it also induces the histopathological characteristics of AD and tau lesions, manifested by NFTs formation, axonal damage, and gliosis. In addition, mononuclear infiltrates without demyelination in the central nervous system (CNS), accompanied by neurological deficits, were observed (such as lameness and limb paralysis). This is the first study showing that immunization against tau may induce tau-like features, and, to circumvent these effects, active immunization is performed using tau fragments or phosphorylated tau fragments[161].
AADvac1 (Axon peptide 108 conjugated to KLH) is the first clinically tested active vaccine against pathologically modified forms of tau protein with an epitope of 294KDNIKHVPGGGS305, the structural determinants essential for pathological tau-tau interactions. Good safety and immunogenicity have been demonstrated in transgenic AD rat models. The produced antibodies are capable of distinguishing between pathological and physiological tau, thus ensuring the specificity against pathological tau protein and significantly delaying tau pathological lesions and deterioration of cognitive behavior[162]. AADvac1 entered phase 1 clinical trials after successful preclinical toxicology and safety studies. The data from the phase 1 trial demonstrate that AADvac1 was safe and well-tolerated, with the majority of participants producing increasing antibody titers after repeat injections[163]. The 18-month open-label extension study showed decreased antibody titers within six months after the last injection, but the intensive dose restored IgG level without treatment-related serious adverse events. Patients with higher IgG responses tended to reduce hippocampal shrinkage and performed better in some cognitive tests[164,165]. In a 24-month phase 2 safety trial, 185 patients with mild to moderate AD were recruited, and its safety and immunogenicity were validated, with more than 95% of immunized participants developing specific tau antibodies and the concentration in CSF averaged 0.3% of serum. Furthermore, some neurodegenerative markers (such as Nfl, pTau217, pTau181, and total tau) were also significantly altered in CSF compared with placebo[166].
ACI-35 is a liposome-based vaccine targeting the pSer396/404 epitope of tau, which activates the immune system and produces antibodies against the pathological conformers of phosphorylated tau protein. ACI-35 has been shown to be safe for long-term injection and can improve clinical status and indices of tau lesions in the brain of P301L mice, without evidence of neuroinflammation or other adverse neurological effects[167]. However, due to its weak immunogenicity, ACI-35.030, the second-generation vaccine of ACI-35, was developed. The redesigned vaccine induced a stronger immune response in rhesus monkeys, and enhanced injection could improve the antibody titer. In July 2019, ACI-35.030 initiated a phase 1b/2a trial in patients with early AD, and the results show that ACI-35.030 generated positive safety, tolerability, and immunogenicity. All participants in the first two dose groups developed anti-tau IgG and IgM responses preferentially targeting phosphorylated tau, with high IgG titers; these results support plans to further advance this vaccine into phase 2/3, where enrollment at the highest dose is currently underway and is expected to be completed in 2023.
Active immunization targeting various forms of tau protein has been proved to reduce tau pathology, and the lasting immune response makes it a promising choice, but the potential autoimmune response is a major concern. Early preclinical reports observed that this problem may be caused by the use of very strong adjuvants not approved for use in humans[161,168].
Passive immunotherapy against tau pathology
Since the effectiveness of isotope immunotherapy was demonstrated in the JNPL3 tau transgenic mouse[169], researchers reported that exogenous monoclonal anti-tau antibodies can effectively reduce tau lesions in the brain, improve cognitive ability, and delay the development of the disease in animal models[101,170]. Compared with active immunization, passive immunization has the ability to reduce the probability of adverse immune response and target specific epitopes, reduce the chance of targeting non-pathological tau, and change the treatment method according to the stage or type of tau lesions to find which tau epitopes are more common; specific IgG isoforms can be selected and changed to improve efficacy. Once diagnostic technology has advanced enough, treatments can be tailored to the individual based on the stage of the disease.
RG7345 ( RO6926496) is a monoclonal antibody specifically targeting the tau phosphoepitope pS422, which is predominantly distributed on dendrites. Phosphorylation of tau at this site is thought to be a pathological form associated with the relocation of tau away from microtubules and toward the soma-dendritic region of neurons[171]. RG7345 enters cells through endocytosis, removes phosphorylated tau protein, and effectively improves the pathological condition of tau[172]. Studies have shown that active vaccination targeting the pS422 tau epitope can induce similar exogenous antibody effects, reduce the level of insoluble pS422 tau, and improve the performance of Thy-Tau22 transgenic mice in the Y maze[173]. Unfortunately, the phase 1 clinical trial of RG7345 lasted less than one year in January 2015, possibly because of poor pharmacokinetics, although no safety or efficacy questions were raised during the trial.
Gosuranemab (BIIB092, BMS-986168, IPN007) is a humanized IgG4 monoclonal antibody targeting extracellular, N-terminal tau fragments (eTau), which are mainly derived from pluripotent stem cells of familial AD (FAD) patients[68,174,175]. Exogenous addition of eTau causes neuronal hyperactivity, which in turn increases Aβ production and neuronal hyperactivity[176]. In mouse models, neutralization of eTau with antibodies reduces Aβ production, effectively improving tau pathological states and patients’ cognitive and behavioral performance[177,178]. In a clinical study in healthy volunteers, infusion caused a dose-dependent increase in blood and CSF of gosuranemab, and a 67%-97% decrease in CSF of unbound eTau after four weeks, without serious or severe adverse events[179]. However, in the gosuranemab phase 2 trial TANGO on patients with mild cognitive impairment caused by AD or mild AD, there was no beneficial effect compared with placebo. On 16 June 2021, TANGO and the development of gosuranemab were terminated.
Semorinemab (RO7105705, MTAU9937A, RG6100) is a monoclonal anti-tau IgG4 antibody targeting the N-terminal region of tau protein, mainly targeting extracellular tau. Semorinemab can bind the N-terminal of all six human tau protein isomers, both tau monomeric and oligomeric, regardless of the phosphorylation state. By binding with tau protein, it can delay their spread between neuron cells. In addition, the purpose of this antibody is to explore antibodies with reducing effector function in an effort to limit the activation of microglia leading to the inflammatory response[180]. Phase 1 clinical trials did not produce serious adverse effects, but also did not produce good results. The phase 2 trial TAURIEL showed that plasma semorinemab was increased in a dose-dependent manner, with a half-life of 32 days, and approximately 0.3% of the antibody entered the CNS. Both CSF total tau and phosphorylated tau decreased, but the total tau value in plasma increased with increasing dose of semorinemab. The antibody did not alter the downstream CSF markers of neurodegeneration and inflammation (such as NFL, neurogranin, S100B, IL-6, and sTREM2) but increased the inflammatory marker YKL-40. YKL-40 protein increases with the development of AD and is associated with brain atrophy and other deleterious effects[181]. Another Phase 2 clinical trial, LAURIET, is underway.
Zagotenemab (LY3303560) is a humanized anti-tau antibody targeting the conformational epitope MC1 (amino acids residues 312-322) of tau, which is an early pathological conformation of tau[182,183]. Zagotenemab selectively binds and neutralizes soluble tau aggregates with high affinity rather than monomers, recognizing a conformational epitope in the N-terminal region of tau protein. In tau transgenic mice, treatment with MC1 reduces phosphorylated tau levels and neurofibrillary pathology, which is mediated by microglia-dependent or neuron-dependent tau/antibody clearance[184,185]. In October 2021, an investor disclosed that a phase 2 trial of zagotenemab in subjects with prodromal to mild AD did not meet its primary endpoint, and the development of the antibody was terminated.
JNJ-63733657 is a humanized IgG1 antibody targeting the microtubule-binding domain (MTBR) of tau protein, which has a high affinity for tau phosphorylated at Thr217 (pT217)[186]. The antibody can more effectively interfere with the transmission of pathogenic and aggregated tau between cells than the antibody against the N-terminal of tau. JNJ-63733657 has been reported to eliminate the “seed” of pathogenic tau in cells and interfere with the dissemination of pathologically aggregated tau proteins in mouse models. A phase 1 clinical trial validated JNJ-63733657 to be safe and tolerable in 72 healthy volunteers. The serum pharmacokinetics was linear with dose and 0.2% ended up in CSF[187]. Single or multiple administrations resulted in a dose-dependent reduction of free p217 tau in CSF[188]. In January 2021, a phase 2 study of 420 patients with early AD symptoms and tau positive PET scan began and is expected to continue until 2025.
Bepranemab (UCB0107, UCB 0107, Antibody D) is a humanized monoclonal IgG4 antibody targeting 235-250 amino acids near the MTBR of tau, which may be more effective for preventing the transmission of pathogenic tau in cell than an antibody targeting N-terminal tau. It is also true that the antibody showed greater efficacy than other antibodies in preventing the seeding and aggregation of pathological tau in tests on cells. Bepranemab can prevent pathologic tau seeds in transgenic mice injected with AD brain extracts and in mice injected with K18 P301L tau fibrils[189,190]. In the clinical trials, no adverse events or safety issues, drug-resistant antibodies, etc., were reported. While serum and CSF concentrations increased in a dose-dependent manner, the CSF/serum ratio remained unchanged at different doses. The experiment is in phase 2 for patients with MCI or mild AD dementia and will run until November 2025.
PNT001 is a monoclonal antibody targeting the cis isomer of phosphorylated tau at threonine 231 (cis-pT231). Cis-pT231-tau is a neurotoxic conformation that has been shown to be one of the main drivers of neurodegenerative diseases, including traumatic brain injury, chronic traumatic encephalopathy, vascular dementia, and AD. Cis-pT231-tau is resistant to dephosphorylation and degradation, promotes tau aggregation, and accelerates neurodegeneration[191,192]. In the human brain, the level of cis-pT231 in CSF correlates with the severity of brain injury[193]. In a mouse model of vascular dementia, PNT001 reduced neurodegeneration and ameliorated cognitive impairment[194]. PNT001 blocks the diffusion of toxic tau protein by accurately targeting and neutralizing tau protein carrying cis-pT231, thereby protecting the normal functioning of the brain and treating neurodegenerative diseases. Phase 1 clinical trial results show that the antibody produced dose-related blood and CSF concentrations that remained constant over 28 days, and it was well tolerated. A study is currently underway on acute traumatic brain injury and is expected to be completed by January 2023.
BIIB076 (NI-105, 6C5 huIgG1/l) is a human recombinant monoclonal IgG1 antibody targeting the mid-domain of tau, which blocks tau aggregation in vitro and tau transmission between neurons[195]. It can recognize monomers, fibrosis, and tau isolated from the brains of healthy subjects and patients with AD. In preclinical studies, following intravenous injection in cynomolgus monkeys, BIIB076 exhibited dose-dependent increases in serum exposures. Total and free tau that were unbound to BIIB076 in CSF decreased significantly, while there were no adverse BIIB076-related toxicology changes or pathology-related findings[196]. These data establish a positive safety profile for BIIB076 for inclusion in a phase 1 trial, which was completed in March 2020. The results show that, as the concentration increased, some side effects occurred, but the safety profile is acceptable. BIIB076 engaged its target, halving the concentration of unbound mid-region-bearing tau in the CSF one week after infusion and the reduction persisted up to three weeks.
Tilavonemab (C2N8E12 /ABBV8E12/ HJ9.3) is a humanized IgG4 antibody targeting 25-30 aa residues at the N-terminal of tau protein, which mainly recognizes aggregated and extracellular pathogenic tau. This antibody cannot enter neuronal cells, and thus only functions outside the cells[101,197,198]. In preclinical studies, the antibody can block tau seeding caused by exogenous tau aggregates and prevent the transneuronal spread of pathological tau[197,198]. The levels of aggregated and hyperphosphorylated tau and brain atrophy can be reduced by injecting antibodies into P301S tau-transgenic mice, thus improving their cognitive performance[101,102]. Although the study found that tilavonemab did not provide more benefits than placebo, phase 2 clinical trials were conducted based on the acceptable safety and tolerability of single-dose tilavonemab to assess the efficacy and safety of multi-dose in early AD or PSP patients[199]. However, the analysis of phase 2 of PSP trial data showed that free tau in CSF was decreased and total tau in plasma increased in the treatment group, but the antibody did not show efficacy[200]. In July 2021, it was announced that the development of tilavonemab was discontinued, and it will be removed from its pipeline.
E2814 is a humanized monoclonal IgG1 antibody targeting the HVPGG sequence that recognizes the second (aa 299-303) and fourth (aa 362-366) repeats of MTBR in tau, which recognizes the 4R and 3R tau isoforms. It is capable of binding extracellular tau, inhibiting the cell-to-cell spread of pathogenic species, and facilitating microglial clearance[201]. In mice injected with K18 P301L tau fibrils, a model of tau transmission, E2814 modestly reduced the propagation of aggregated tau. Some data suggest that tau antibodies of the mid-domain are more effective at interfering with the spread of pathogenic, aggregated tau than N-terminally targeted anti-tau antibodies currently in clinical trials. A phase 1 trial testing the safety and tolerability of a single intravenous infusion in healthy adults showed no significant drug-related clinical changes or dose-limiting events, but there was a dose-related increase in antibody-tau association, which persisted for at least a month. In 2021, a multiple-ascending-dose trial was conducted to detect E2814’s safety, pharmacokinetics, and induction of anti-E2814 antibodies. The trial will run until November 2022. Meanwhile, E2814 was selected for evaluation in patients with pathogenic APP and presenilin mutations in the DIAN-TU prophylaxis trial, which is expected to be completed in April 2024.
Lu AF87908 is a humanized mouse IgG1 monoclonal antibody produced by immunization of mice with the tau386-408 epitope and phosphorylating at Ser396 and Ser404. This antibody preferentially binds aggregates of hyperphosphorylated tau and reduces the ability of brain-derived tau to seed aggregates in cultured neurons and rTg4510 tau transgenic mice[202], and it can mediate the uptake and lysosomal degradation of tau aggregates in microglia via the antibody’s Fcγ receptor[203]. A phase 1 study is currently underway to test its safety, tolerability, and pharmacokinetics in healthy individuals and AD patients. The study is expected to be completed by July 2022.
CURRENT CHALLENGES AND FUTURE OPPORTUNITIES IN TAU-TARGETING IMMUNOTHERAPY
Challenges in tau immunotherapy
Currently, research on a targeted tau vaccine is still in its infancy. Many unknown fields, such as the amino acid sequence of tau protein suitable for synthetic vaccine, methods to determine vaccine effectiveness, and the dose and mode of vaccination, require further exploration. In the meantime, the following issues must be considered in immunotherapy research targeting tau protein.
BBB crossing
The reason for the failure of most studies is the inability of drugs to cross the BBB. Increasing the concentration of therapeutic drugs in the brain can not only improve the efficacy of antibodies, so as to improve the success rate of clinical trials, but also reduce the production and treatment costs and the number of treatments required. Therefore, it is necessary to strengthen the delivery of antibodies and other biological therapeutic drugs used to treat central nervous system diseases.
Nanotechnology has recently emerged as a promising route for cross-BBB drug delivery to the CNS. The continuous and accurate drug targeting ability of nanoparticles (NPs) can make up for the defects of insufficient drug concentration in the CSF. Ideal NPs should have appropriate physiochemical properties, good biocompatibility, and effective production magnification[204]. NPs are small, nanoscale carrier structures that can be designed or modified to encapsulate or attach to molecules, peptides, proteins, antibodies, and nucleic acids[205]. There is currently a variety of NPs being explored for CNS drug delivery, including gold, hydrogels, liposomes, etc.[206]. The use of different nanocarriers is currently one of the most widely used state-of-the-art technologies to overcome the shortcomings of existing drugs and provide new therapeutic approaches, particularly in diseases where access to affected organs is difficult. Currently, some NPs targeting tau have been developed. Ghalandari et al. prepared folic acid functionalized gold NPs (FA-AuNPs) and gold-shelled Fe3O4 NPs (AuFeNPs), which showed a binding affinity for tubulin and tau[207]. Protein-capped metal (PC-Fe3O4 and PC-CdS) NPs present a novel function, which act as a powerful tau aggregation inhibitor in AD[208]. Cur-loaded T807/RPCNP NPs not only effectively penetrate the BBB but also selectively combine with p-tau with high affinity, thereby inhibiting the important pathways in tau-related AD pathogenesis and alleviating the memory impairment of AD mouse models[204]. These NPs have become a promising treatment strategy for AD through their high bioavailability in the brain. However, due to technical and cost limitations, as well as exhibiting long-term toxicity, only a few nanoparticles targeting diseases of the CNS are currently in clinical trials[209,210]. Another delivery system, the exosome, has also become a hot topic of current research, as its lipid bilayer reduces renal clearance, encapsulates and protects drugs that will be degraded, and allows the recipient cells to recognize and absorb the homing properties of vesicles, and its low immunogenicity makes it a promising drug delivery system[211]. Studies have shown that exosomes loaded with drugs can cross the BBB when injected intravenously in mice[211,212]. Advances in these technologies for medical drug development, drug delivery, and novel diagnostic biomarkers may serve as promising option for the treatment and diagnosis of AD as well as other neurodegenerative diseases.
Cell membrane crossing
Tau is mainly concentrated in neuronal cells, and how antibodies cross through membranes has become a major challenge hindering antibody development and application. Previous studies have shown that many antibodies can actually function intra/extracellularly[213,214] and are detectable in neurons[213,215,216], although some antibodies are not taken up into neurons[213,217].
Tau antibodies can cross the BBB and enter neurons, which are then mediated by the FcR or endocytosis[218,219]. Intracellularly, antibodies can bind to tau aggregates within the endosomal-lysosomal system and promote their breakdown, thereby increasing the pathway for lysosomal enzymes to degrade the aggregates[216], isolating tau assemblies in the cytoplasm and preventing their release from neurons, or promoting proteasomal degradation through the binding of the E3 ubiquitin-protein ligase TRIM21[220].
Of course, there is also a small amount of tau outside the cell. Extracellular tau is therefore crucial in the spread of tau, and extracellular antibodies can change the process of the disease by preventing the transmission of pathological tau. Extracellular antibodies act mainly by isolating tau aggregates, interfering with their assembly, and promoting phagocytosis of microglia, thus blocking the spread of tau pathology among neurons[184,197] [Figure 3].
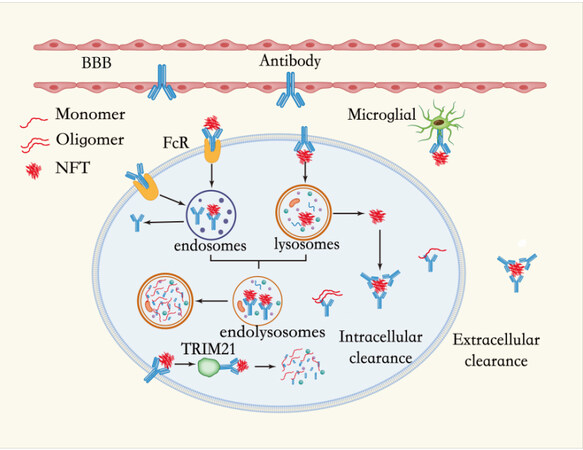
Figure 3. Mechanism of action of tau protein clearance. (1) Intracellular clearance: antibodies can cross the BBB and enter the neuron, and then enter the cell either mediated by FcR or through endocytosis. Intracellularly, antibodies can bind to tau aggregates within the endosomal-lysosomal system and promote their breakdown, thereby increasing the pathway for lysosomal enzymes to degrade the aggregates while isolating tau assemblies in the cytoplasm and preventing their release from neurons or promoting proteasomal degradation through the binding of the E3 ubiquitin-protein ligase TRIM21. (2) Extracellular clearance: extracellular antibodies act to block the spread of tau pathology between neurons by isolating tau aggregates, interfering with their assembly, and promoting phagocytosis by microglia.
Vaccine biosafety
Studies have shown that some of the phosphatase sites targeted by immunotherapy are presented in the brains of healthy people. This has raised concerns about the safety of vaccines targeting specific phosphorylation sites. Concurrently, these treatments have been studied by selectively targeting only one or two phosphate sites out of the 45 phosphate sites identified in AD brains. However, it is not known whether all phosphorylation epitopes of pathological tau coexist on the same tau molecule, or whether different pathological tau subspecies display a variety of different phosphorylation sites.
At the same time, compared to Aβ, tau is a much larger antigen, so it is very important to select the correct immunogenic epitopes. Previously, targeting the N-terminus of the tau protein was a hot research topic because of its ability to generate high-affinity antibodies. However, thus far, antibodies that bind to this region appear to have had little success in phase 2 trials. Therefore, antibodies against tau mid-region, especially MTBR, are an important target in recent studies. Several antibodies currently targeting this epitope suggest that mid-region antibodies are more effective at interfering with the spread of pathogenic, aggregated tau than anti-tau antibodies targeting the N-terminus. The reason is that the region can drive tau aggregation, and the antibody binding to this site can prevent the spread of misfolded forms of tau[221].
Future directions
Currently, clinical trials of tau-based drugs aimed at gain-of-toxic-tau function (e.g., dysregulation of PTMs and tau aggregation) or loss-of-function (microtubules instability) have been increasingly conducted in tauopathy patients. In addition, as the understanding of tau proteins increases, more and more studies targeting tau are being conducted.
Biomolecular condensation of intrinsically disordered proteins/regions (IDPs/IDRs) via liquid-liquid phase separation (LLPS) has gained widespread interest as the rapidly unfolding role of phase-separated condensates in a variety of cellular functions and human diseases[222]. LLPS of tau is essential to regulate some cellular physiological functions; however, abnormal phase separation can lead to the transition of soluble tau to fibrotic tau, and the generation of phase transition droplets provides local high protein concentration and positive charge density for the generation of fibrillar amyloid structures, favoring the recruitment of polyanions (ribonucleic acid, heparin, etc.) involved in the formation of neurofibrillary tangles[223]. The rapid development of the LLPS mechanism has undoubtedly changed our understanding of various biological activities and disease conditions. The basic research on LLPS and human diseases will continue to be improved and translated into clinical practice.
apoE4 is the strongest genetic risk factor for late-onset AD, which is found in some neurons with NFTs, and the specific interaction of apoE with tau may regulate tau metabolism in neurons and alter the rate of PHF and NFT formation[224]. Removal of apoE4 substantially reduces tau-regulated neurodegeneration and decreases p-tau lesions, tau-induced synaptic loss, and phagocytosis of synaptic elements by microglia[225]. In the absence of microglia, the pathological progression of p-tau is prevented and the pathogenesis of p-tau is also largely driven by microglia-mediated inflammation[226]. Overexpression of the apoE metabolic receptor, low-density lipoprotein receptor, in P301S tauopathy mice significantly reduces brain apoE and improves tau pathology and neurodegeneration[227]. These results suggest that an in-depth understanding of the mechanisms of action of tau protein will provide novel therapeutic approaches for AD.
In brief, targeting tau is a promising treatment strategy. However, various challenges, including BBB restricting the entry of drugs and reducing the effective drug concentration, the lack of selectivity and specificity of target proteins leading to insufficient or ineffective treatment effect, or the difference of phenotype between animal models and humans resulting in the failure of clinical transformation of drugs, as well as the selection of disease treatment stages, have limited the development of drugs. With the improvement of diagnostic methods, the intervention time was transferred to MCI stage or early prodromal stage of AD, and treatment should be carried out before symptoms appear. The development of early diagnosis methods allows for diagnosis and treatment early for diseases. Active immunotherapy can prevent the disease in the early stage but may lack the corresponding specificity and produce autoimmune side effects. Once the disease progresses and a large number of pathological proteins appear, a single treatment cannot be enough to alleviate the disease progress. The combination of passive immunotherapy and various treatment methods such as inhibiting its aggregation, transmission, protein expression, etc. will improve the effect of treatment to a certain extent. Although many candidates targeting tau have been discovered and developed at present, few are really useful in the clinic. Further continuing basic research on the underlying causes and mechanisms of dysfunction in AD is critical to identifying new targets for intervention.
DECLARATIONS
Authors’ contributionsResearch and drafting of the article: Guo Y, Tan J
Coordinate the first draft: Li S, Zeng LH
Edit the article: Li S, Tan J
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis study was supported by the High-level Talent Foundation of Guizhou Medical University (YJ19017, HY2020, J.T.), National Natural Science Foundation of China (NSFC) (82171423, 82060211, J.T.).
Conflicts of interestNot applicable.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
2. Wang QH, Wang X, Bu XL, et al. Comorbidity burden of dementia: a hospital-based retrospective study from 2003 to 2012 in seven cities in China. Neurosci Bull 2017;33:703-10.
4. Busche MA, Hyman BT. Synergy between amyloid-β and tau in Alzheimer’s disease. Nat Neurosci 2020;23:1183-93.
5. Hernandez P, Lee G, Sjoberg M, Maccioni RB. Tau phosphorylation by cdk5 and Fyn in response to amyloid peptide Abeta (25-35): involvement of lipid rafts. J Alzheimers Dis 2009;16:149-56.
6. Terwel D, Muyllaert D, Dewachter I, et al. Amyloid activates GSK-3beta to aggravate neuronal tauopathy in bigenic mice. Am J Pathol 2008;172:786-98.
7. Gamblin TC, Chen F, Zambrano A, et al. Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer’s disease. Proc Natl Acad Sci U S A 2003;100:10032-7.
8. Peters F, Salihoglu H, Pratsch K, et al. Tau deletion reduces plaque-associated BACE1 accumulation and decelerates plaque formation in a mouse model of Alzheimer’s disease. EMBO J 2019;38:e102345.
9. Pascoal TA, Mathotaarachchi S, Mohades S, et al. Amyloid-β and hyperphosphorylated tau synergy drives metabolic decline in preclinical Alzheimer’s disease. Mol Psychiatry 2017;22:306-11.
10. Fortea J, Vilaplana E, Alcolea D, et al. Alzheimer’s disease neuroimaging initiative. Cerebrospinal fluid β-amyloid and phospho-tau biomarker interactions affecting brain structure in preclinical Alzheimer disease. Ann Neurol 2014;76:223-30.
11. Timmers M, Tesseur I, Bogert J, et al. Relevance of the interplay between amyloid and tau for cognitive impairment in early Alzheimer’s disease. Neurobiol Aging 2019;79:131-41.
12. Kabir MT, Uddin MS, Mamun AA, et al. Combination drug therapy for the management of Alzheimer’s disease. Int J Mol Sci 2020;21:3272.
14. Hampel H, Vassar R, De Strooper B, et al. The β-Secretase BACE1 in Alzheimer’s disease. Biol Psychiatry 2021;89:745-56.
15. Penninkilampi R, Brothers HM, Eslick GD. Pharmacological agents targeting γ-secretase increase risk of cancer and cognitive decline in Alzheimer’s disease patients: a systematic review and meta-analysis. J Alzheimers Dis 2016;53:1395-404.
16. Wolfe MS. Probing mechanisms and therapeutic potential of γ-Secretase in Alzheimer’s disease. Molecules 2021;26:388.
17. Sagnou M, Mavroidi B, Kaminari A, Boukos N, Pelecanou M. Novel isatin thiosemicarbazone derivatives as potent inhibitors of β-Amyloid peptide aggregation and toxicity. ACS Chem Neurosci 2020;11:2266-76.
18. Sterner RM, Takahashi PY, Yu Ballard AC. Active vaccines for Alzheimer disease treatment. J Am Med Dir Assoc 2016;17:862.e11-5.
19. Gilman S, Koller M, Black RS, et al. AN1792(QS-21)-201 Study Team. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology 2005;64:1553-62.
20. La Joie R, Visani AV, Baker SL, et al. Prospective longitudinal atrophy in Alzheimer’s disease correlates with the intensity and topography of baseline tau-PET. Sci Transl Med 2020;12:eaau5732.
21. Kametani F, Hasegawa M. Reconsideration of amyloid hypothesis and Tau hypothesis in Alzheimer’s disease. Front Neurosci 2018;12:25.
22. Pedersen JT, Sigurdsson EM. Tau immunotherapy for Alzheimer’s disease. Trends Mol Med 2015;21:394-402.
24. Tapia-Rojas C, Cabezas-Opazo F, Deaton CA, Vergara EH, Johnson GVW, Quintanilla RA. It’s all about tau. Prog Neurobiol 2019;175:54-76.
25. Ait-Bouziad N, Chiki A, Limorenko G, Xiao S, Eliezer D, Lashuel HA. Phosphorylation of the overlooked tyrosine 310 regulates the structure, aggregation, and microtubule- and lipid-binding properties of Tau. J Biol Chem 2020;295:7905-22.
26. Braak H, Braak E. Staging of alzheimer’s disease-related neurofibrillary changes. Neurobiol Aging 1995;16:271-8.
27. Clavaguera F, Duyckaerts C, Haïk S. Prion-like properties of Tau assemblies. Curr Opin Neurobiol 2020;61:49-57.
28. Olsson B, Lautner R, Andreasson U, et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: a systematic review and meta-analysis. Lancet Neurol 2016;15:673-84.
29. Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 2006;112:389-404.
30. Alquezar C, Arya S, Kao AW. Tau post-translational modifications: dynamic transformers of Tau function, degradation, and aggregation. Front Neurol 2020;11:595532.
31. Köpke E, Tung YC, Shaikh S, et al. Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J Biol Chem 1993;268:24374-84.
32. Noble W, Hanger DP, Miller CC, Lovestone S. The importance of tau phosphorylation for neurodegenerative diseases. Front Neurol 2013;4:83.
33. Alonso A, Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc Natl Acad Sci U S A 2001;98:6923-8.
34. Caballero B, Bourdenx M, Luengo E, et al. Acetylated tau inhibits chaperone-mediated autophagy and promotes tau pathology propagation in mice. Nat Commun 2021;12:2238.
35. Min SW, Cho SH, Zhou Y, et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 2010;67:953-66.
36. Cohen TJ, Guo JL, Hurtado DE, et al. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat Commun 2011;2:252.
37. Wang JZ, Grundke-Iqbal I, Iqbal K. Glycosylation of microtubule-associated protein tau: an abnormal posttranslational modification in Alzheimer’s disease. Nat Med 1996;2:871-5.
38. Reynolds MR, Berry RW, Binder LI. Site-specific nitration and oxidative dityrosine bridging of the tau protein by peroxynitrite: implications for Alzheimer’s disease. Biochemistry 2005;44:1690-700.
39. Cotman CW, Poon WW, Rissman RA, Blurton-Jones M. The role of caspase cleavage of tau in Alzheimer disease neuropathology. J Neuropathol Exp Neurol 2005;64:104-12.
40. Zhao X, Kotilinek LA, Smith B, et al. Caspase-2 cleavage of tau reversibly impairs memory. Nat Med 2016;22:1268-76.
41. Berry RW, Abraha A, Lagalwar S, et al. Inhibition of tau polymerization by its carboxy-terminal caspase cleavage fragment. Biochemistry 2003;42:8325-31.
42. Pérez MJ, Vergara-Pulgar K, Jara C, Cabezas-Opazo F, Quintanilla RA. Caspase-cleaved Tau impairs mitochondrial dynamics in Alzheimer’s disease. Mol Neurobiol 2018;55:1004-18.
43. Lee MJ, Lee JH, Rubinsztein DC. Tau degradation: the ubiquitin-proteasome system versus the autophagy-lysosome system. Prog Neurobiol 2013;105:49-59.
44. Funk KE, Thomas SN, Schafer KN, et al. Lysine methylation is an endogenous post-translational modification of tau protein in human brain and a modulator of aggregation propensity. Biochem J 2014;462:77-88.
45. Maina MB, Al-Hilaly YK, Oakley S, et al. Dityrosine cross-links are present in Alzheimer’s disease-derived tau oligomers and paired helical filaments (PHF) which promotes the stability of the PHF-core tau (297-391). bioRxiv 2022; doi: 10.1101/2022.05.28.493839.
46. Maina MB, Al-Hilaly YK, Burra G, et al. Oxidative stress conditions result in trapping of PHF-core Tau (297-391) intermediates. Cells 2021;10:703.
47. Lasagna-Reeves CA, Castillo-Carranza DL, Guerrero-Muoz MJ, Jackson GR, Kayed R. Preparation and characterization of neurotoxic tau oligomers. Biochemistry 2010;49:10039-41.
48. Qian W, Shi J, Yin X, et al. PP2A regulates tau phosphorylation directly and also indirectly via activating GSK-3beta. J Alzheimers Dis 2010;19:1221-9.
49. Nicolia V, Fuso A, Cavallaro RA, Di Luzio A, Scarpa S. B vitamin deficiency promotes tau phosphorylation through regulation of GSK3beta and PP2A. J Alzheimers Dis 2010;19:895-907.
50. Lauretti E, Dincer O, Praticò D. Glycogen synthase kinase-3 signaling in Alzheimer’s disease. Biochim Biophys Acta Mol Cell Res 2020;1867:118664.
51. Serenó L, Coma M, Rodríguez M, et al. A novel GSK-3beta inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo. Neurobiol Dis 2009;35:359-67.
52. Wang H, Huang S, Yan K, et al. Tideglusib, a chemical inhibitor of GSK3β, attenuates hypoxic-ischemic brain injury in neonatal mice. Biochim Biophys Acta 2016;1860:2076-85.
53. Morales-Garcia JA, Luna-Medina R, Alonso-Gil S, et al. Glycogen synthase kinase 3 inhibition promotes adult hippocampal neurogenesis in vitro and in vivo. ACS Chem Neurosci 2012;3:963-71.
54. Lovestone S, Boada M, Dubois B, et al. ARGO investigators. A phase II trial of tideglusib in Alzheimer’s disease. J Alzheimers Dis 2015;45:75-88.
55. Matsunaga S, Fujishiro H, Takechi H. Efficacy and safety of glycogen synthase kinase 3 inhibitors for Alzheimer’s disease: A Systematic Review and Meta-Analysis. J Alzheimers Dis 2019;69:1031-9.
56. Hampel H, Lista S, Mango D, et al. Alzheimer Precision Medicine Initiative (APMI). Lithium as a treatment for Alzheimer’s disease: the systems pharmacology perspective. J Alzheimers Dis 2019;69:615-29.
57. Smith KA, Cipriani A. Lithium and suicide in mood disorders: updated meta-review of the scientific literature. Bipolar Disord 2017;19:575-86.
58. Hampel H, Ewers M, Bürger K, et al. Lithium trial in Alzheimer’s disease: a randomized, single-blind, placebo-controlled, multicenter 10-week study. J Clin Psychiatry 2009;70:922-31.
59. Forlenza OV, Diniz BS, Radanovic M, Santos FS, Talib LL, Gattaz WF. Disease-modifying properties of long-term lithium treatment for amnestic mild cognitive impairment: randomised controlled trial. Br J Psychiatry 2011;198:351-6.
60. Forlenza OV, Radanovic M, Talib LL, Gattaz WF. Clinical and biological effects of long-term lithium treatment in older adults with amnestic mild cognitive impairment: randomised clinical trial. Br J Psychiatry 2019;215:668-74.
61. Nunes MA, Viel TA, Buck HS. Microdose lithium treatment stabilized cognitive impairment in patients with Alzheimer’s disease. Curr Alzheimer Res 2013;10:104-7.
62. Damri O, Shemesh N, Agam G. Is there justification to treat neurodegenerative disorders by repurposing drugs? Int J Mol Sci 2020;22:189.
63. Morris G, Berk M. The putative use of lithium in Alzheimer’s disease. Curr Alzheimer Res 2016;13:853-61.
64. Gildengers A, Aizenstein H, Anderson S, et al. Lithium as a treatment to prevent impairment of cognition in elders(LATTICE)2017;Available from: https://clinicaltrials.gov/ct2/show/NCT03185208 [Last accessed on 20 Jul 2022].
65. Georgievska B, Sandin J, Doherty J, et al. AZD1080, a novel GSK3 inhibitor, rescues synaptic plasticity deficits in rodent brain and exhibits peripheral target engagement in humans. J Neurochem 2013;125:446-56.
66. Hu S, Hu M, Liu J, et al. Phosphorylation of Tau and α-synuclein induced neurodegeneration in MPTP mouse model of Parkinson’s disease. Neuropsychiatr Dis Treat 2020;16:651-63.
67. Cruz JC, Tseng H, Goldman JA, Shih H, Tsai L. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron 2003;40:471-83.
68. Iqbal K, Liu F, Gong CX. Tau and neurodegenerative disease: the story so far. Nat Rev Neurol 2016;12:15-27.
69. Piedrahita D, Hernández I, López-Tobón A, et al. Silencing of CDK5 reduces neurofibrillary tangles in transgenic alzheimer’s mice. J Neurosci 2010;30:13966-76.
70. Zhao Y, Wang C, He W, Cai Z. Ameliorating Alzheimer’s-like pathology by Minocycline via inhibiting Cdk5/p25 signaling. Curr Neuropharmacol 2021; doi: 10.2174/1570159X19666211202124925.
71. Li T, Hawkes C, Qureshi HY, Kar S, Paudel HK. Cyclin-dependent protein kinase 5 primes microtubule-associated protein tau site-specifically for glycogen synthase kinase 3beta. Biochemistry 2006;45:3134-45.
72. Engmann O, Giese KP. Crosstalk between Cdk5 and GSK3beta: implications for Alzheimer’s disease. Front Mol Neurosci 2009;2:2.
73. Wen Y, Planel E, Herman M, et al. Interplay between cyclin-dependent kinase 5 and glycogen synthase kinase 3 beta mediated by neuregulin signaling leads to differential effects on tau phosphorylation and amyloid precursor protein processing. J Neurosci 2008;28:2624-32.
74. Wang HH, Li Y, Li A, et al. Forskolin induces hyperphosphorylation of Tau accompanied by cell cycle reactivation in primary hippocampal neurons. Mol Neurobiol 2018;55:696-706.
75. Villa V, Montalto G, Caudano F, Fedele E, Ricciarelli R. Selective inhibition of phosphodiesterase 4D increases tau phosphorylation at Ser214 residue. Biofactors 2022; doi: 10.1002/biof.1847.
76. Zheng-Fischhöfer Q, Biernat J, Mandelkow EM, Illenberger S, Godemann R, Mandelkow E. Sequential phosphorylation of Tau by glycogen synthase kinase-3beta and protein kinase A at Thr212 and Ser214 generates the Alzheimer-specific epitope of antibody AT100 and requires a paired-helical-filament-like conformation. Eur J Biochem 1998;252:542-52.
77. Domise M, Didier S, Marinangeli C, et al. AMP-activated protein kinase modulates tau phosphorylation and tau pathology in vivo. Sci Rep 2016;6:26758.
78. Vingtdeux V, Davies P, Dickson DW, Marambaud P. AMPK is abnormally activated in tangle- and pre-tangle-bearing neurons in Alzheimer’s disease and other tauopathies. Acta Neuropathol 2011;121:337-49.
79. Wang L, Li N, Shi FX, et al. Upregulation of AMPK ameliorates Alzheimer’s disease-like Tau pathology and memory impairment. Mol Neurobiol 2020;57:3349-61.
80. Zhang S, Wang C, Lu J, et al. In-cell NMR study of Tau and MARK2 phosphorylated Tau. Int J Mol Sci 2018;20:90.
81. Wu PR, Tsai PI, Chen GC, et al. DAPK activates MARK1/2 to regulate microtubule assembly, neuronal differentiation, and tau toxicity. Cell Death Differ 2011;18:1507-20.
82. Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci 2005;22:1942-50.
83. Virshup DM, Shenolikar S. From promiscuity to precision: protein phosphatases get a makeover. Mol Cell 2009;33:537-45.
84. Wang JZ, Grundke-Iqbal I, Iqbal K. Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. Eur J Neurosci 2007;25:59-68.
85. van Eersel J, Ke YD, Liu X, et al. Sodium selenate mitigates tau pathology, neurodegeneration, and functional deficits in Alzheimer’s disease models. Proc Natl Acad Sci U S A 2010;107:13888-93.
86. Malpas CB, Vivash L, Genc S, et al. A phase IIa randomized control trial of VEL015 (Sodium Selenate) in mild-moderate Alzheimer’s disease. J Alzheimers Dis 2016;54:223-32.
87. Vivash L, Malpas CB, Hovens CM, et al. Sodium selenate as a disease-modifying treatment for mild-moderate Alzheimer’s disease: an open-label extension study. BMJ Neurol Open 2021;3:e000223.
88. Gong CX, Liu F, Grundke-Iqbal I, Iqbal K. Post-translational modifications of tau protein in Alzheimer’s disease. J Neural Transm (Vienna) 2005;112:813-38.
89. Liu F, Iqbal K, Grundke-Iqbal I, Hart GW, Gong CX. O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer’s disease. Proc Natl Acad Sci U S A 2004;101:10804-9.
90. Graham DL, Gray AJ, Joyce JA, et al. Increased O-GlcNAcylation reduces pathological tau without affecting its normal phosphorylation in a mouse model of tauopathy. Neuropharmacology 2014;79:307-13.
91. Hastings NB, Wang X, Song L, et al. Inhibition of O-GlcNAcase leads to elevation of O-GlcNAc tau and reduction of tauopathy and cerebrospinal fluid tau in rTg4510 mice. Mol Neurodegener 2017;12:39.
92. Yuzwa SA, Shan X, Macauley MS, et al. Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nat Chem Biol 2012;8:393-9.
93. Gong CX, Liu F, Grundke-Iqbal I, Iqbal K. Impaired brain glucose metabolism leads to Alzheimer neurofibrillary degeneration through a decrease in tau O-GlcNAcylation. J Alzheimers Dis 2006;9:1-12.
94. Zhu Y, Shan X, Yuzwa SA, Vocadlo DJ. The emerging link between O-GlcNAc and Alzheimer disease. J Biol Chem 2014;289:34472-81.
95. Paul S, Haskali MB, Liow JS, et al. Evaluation of a PET radioligand to image O-GlcNAcase in brain and periphery of rhesus monkey and knock-out mouse. J Nucl Med 2019;60:129-34.
96. Shin MK, Vázquez-Rosa E, Koh Y, et al. Reducing acetylated tau is neuroprotective in brain injury. Cell 2021;184:2715-2732.e23.
97. Min SW, Chen X, Tracy TE, et al. Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat Med 2015;21:1154-62.
98. VandeVrede L, Dale ML, Fields S, et al. Open-label phase 1 futility studies of salsalate and young plasma in progressive supranuclear palsy. Mov Disord Clin Pract 2020;7:440-7.
99. Gu J, Xu W, Jin N, et al. Truncation of Tau selectively facilitates its pathological activities. J Biol Chem 2020;295:13812-28.
100. Šimić G, Babić Leko M, Wray S, et al. Tau protein hyperphosphorylation and aggregation in Alzheimer’s disease and other tauopathies, and possible neuroprotective strategies. Biomolecules 2016;6:6.
101. Yanamandra K, Kfoury N, Jiang H, et al. Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron 2013;80:402-14.
102. Yanamandra K, Jiang H, Mahan TE, et al. Anti-tau antibody reduces insoluble tau and decreases brain atrophy. Ann Clin Transl Neurol 2015;2:278-88.
103. Corsetti V, Borreca A, Latina V, et al. Passive immunotherapy for N-truncated tau ameliorates the cognitive deficits in two mouse Alzheimer’s disease models. Brain Commun 2020;2:fcaa039.
104. Singh G, Liu P, Yao KR, et al. Caspase-2 inhibitor blocks Tau truncation and restores excitatory neurotransmission in neurons modeling FTDP-17 tauopathy. ACS Chem Neurosci 2022;13:1549-57.
105. Brier MR, Gordon B, Friedrichsen K, et al. Tau and Aβ imaging, CSF measures, and cognition in Alzheimer’s disease. Sci Transl Med 2016;8:338ra66.
106. Penke B, Szűcs M, Bogár F. Oligomerization and conformational change turn monomeric β-Amyloid and Tau proteins toxic: their role in Alzheimer’s pathogenesis. Molecules 2020;25:1659.
107. Wischik CM, Edwards PC, Lai RY, Roth M, Harrington CR. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc Natl Acad Sci U S A 1996;93:11213-8.
108. Riha PD, Rojas JC, Gonzalez-Lima F. Beneficial network effects of methylene blue in an amnestic model. Neuroimage 2011;54:2623-34.
109. Wischik CM, Staff RT, Wischik DJ, et al. Tau aggregation inhibitor therapy: an exploratory phase 2 study in mild or moderate Alzheimer’s disease. J Alzheimers Dis 2015;44:705-20.
110. Gauthier S, Feldman HH, Schneider LS, et al. Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: a randomised, controlled, double-blind, parallel-arm, phase 3 trial. The Lancet 2016;388:2873-84.
111. Baddeley TC, McCaffrey J, Storey JM, et al. Complex disposition of methylthioninium redox forms determines efficacy in tau aggregation inhibitor therapy for Alzheimer’s disease. J Pharmacol Exp Ther 2015;352:110-8.
112. Wilcock GK, Gauthier S, Frisoni GB, et al. Potential of low dose leuco-methylthioninium bis(Hydromethanesulphonate) (LMTM) monotherapy for treatment of mild Alzheimer’s disease: cohort analysis as modified primary outcome in a phase III clinical trial. J Alzheimers Dis 2018;61:435-57.
113. Kroth H, Ansaloni A, Varisco Y, et al. Discovery and structure activity relationship of small molecule inhibitors of toxic β-amyloid-42 fibril formation. J Biol Chem 2012;287:34786-800.
114. Poli S. SMALL MOLECULES TARGETING TAU PROPAGATION DEMONSTRATE EFFICACY IN AN AGGRESSIVE TAUOPATHY MOUSE MODE. Available from: https://www.acimmune.com/ [Last accessed on 20 Jul 2022].
115. Frautschy SA, Cole GM. Why pleiotropic interventions are needed for Alzheimer’s disease. Mol Neurobiol 2010;41:392-409.
116. Chen M, Du ZY, Zheng X, Li DL, Zhou RP, Zhang K. Use of curcumin in diagnosis, prevention, and treatment of Alzheimer’s disease. Neural Regen Res 2018;13:742-52.
117. Ege D. Action mechanisms of curcumin in Alzheimer’s disease and its brain targeted delivery. Materials (Basel) 2021;14:3332.
118. Prior M, Dargusch R, Ehren JL, Chiruta C, Schubert D. The neurotrophic compound J147 reverses cognitive impairment in aged Alzheimer’s disease mice. Alzheimers Res Ther 2013;5:25.
119. Viswanathan GK, Shwartz D, Losev Y, et al. Purpurin modulates Tau-derived VQIVYK fibrillization and ameliorates Alzheimer’s disease-like symptoms in animal model. Cell Mol Life Sci 2020;77:2795-813.
120. Shin SJ, Park YH, Jeon SG, et al. Red ginseng inhibits Tau aggregation and promotes Tau dissociation in vitro. Oxid Med Cell Longev 2020;2020:7829842.
121. Thapa A, Jett SD, Chi EY. Curcumin attenuates amyloid-β aggregate toxicity and modulates amyloid-β aggregation pathway. ACS Chem Neurosci 2016;7:56-68.
122. Ramesh M, Acharya A, Murugan NA, Ila H, Govindaraju T. Thiophene-based dual modulators of Aβ and Tau aggregation. Chembiochem 2021;22:3348-57.
123. Son SH, Do JM, Yoo JN, et al. Identification of ortho catechol-containing isoflavone as a privileged scaffold that directly prevents the aggregation of both amyloid β plaques and tau-mediated neurofibrillary tangles and its in vivo evaluation. Bioorg Chem 2021;113:105022.
124. DeVos SL, Miller RL, Schoch KM, et al. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci Transl Med 2017;9:eaag0481.
125. DeVos SL, Goncharoff DK, Chen G, et al. Antisense reduction of tau in adult mice protects against seizures. J Neurosci 2013;33:12887-97.
126. Biswas S, Kalil K. The microtubule-associated protein Tau mediates the organization of microtubules and their dynamic exploration of actin-rich lamellipodia and filopodia of cortical growth cones. J Neurosci 2018;38:291-307.
127. Dawson HN, Ferreira A, Eyster MV, Ghoshal N, Binder LI, Vitek MP. Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J Cell Sci 2001;114:1179-87.
128. Yu W, Qiang L, Solowska JM, Karabay A, Korulu S, Baas PW. The microtubule-severing proteins spastin and katanin participate differently in the formation of axonal branches. Mol Biol Cell 2008;19:1485-98.
129. Velazquez R, Ferreira E, Tran A, et al. Acute tau knockdown in the hippocampus of adult mice causes learning and memory deficits. Aging Cell 2018;17:e12775.
130. Sapir T, Frotscher M, Levy T, Mandelkow EM, Reiner O. Tau’s role in the developing brain: implications for intellectual disability. Hum Mol Genet 2012;21:1681-92.
131. Violet M, Delattre L, Tardivel M, et al. A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Front Cell Neurosci 2014;8:84.
132. Ikegami S. Muscle weakness, hyperactivity, and impairment in fear conditioning in tau-deficient mice. Neurosci Lett 2000;279:129-32.
133. Barbier P, Zejneli O, Martinho M, et al. Role of Tau as a microtubule-associated protein: structural and functional aspects. Front Aging Neurosci 2019;11:204.
134. Paonessa F, Evans LD, Solanki R, et al. Microtubules deform the nuclear membrane and disrupt nucleocytoplasmic transport in Tau-mediated frontotemporal dementia. Cell Rep 2019;26:582-593.e5.
135. Ivashko-Pachima Y, Sayas CL, Malishkevich A, Gozes I. ADNP/NAP dramatically increase microtubule end-binding protein-Tau interaction: a novel avenue for protection against tauopathy. Mol Psychiatry 2017;22:1335-44.
136. Varidaki A, Hong Y, Coffey ET. Repositioning microtubule stabilizing drugs for brain disorders. Front Cell Neurosci 2018;12:226.
137. Vulih-Shultzman I, Pinhasov A, Mandel S, et al. Activity-dependent neuroprotective protein snippet NAP reduces tau hyperphosphorylation and enhances learning in a novel transgenic mouse model. J Pharmacol Exp Ther 2007;323:438-49.
138. Matsuoka Y, Jouroukhin Y, Gray AJ, et al. A neuronal microtubule-interacting agent, NAPVSIPQ, reduces tau pathology and enhances cognitive function in a mouse model of Alzheimer’s disease. J Pharmacol Exp Ther 2008;325:146-53.
139. Merenlender-Wagner A, Pikman R, Giladi E, Andrieux A, Gozes I. NAP (davunetide) enhances cognitive behavior in the STOP heterozygous mouse--a microtubule-deficient model of schizophrenia. Peptides 2010;31:1368-73.
140. Morimoto BH, Schmechel D, Hirman J, Blackwell A, Keith J, Gold M. AL-108-211 Study. A double-blind, placebo-controlled, ascending-dose, randomized study to evaluate the safety, tolerability and effects on cognition of AL-108 after 12 weeks of intranasal administration in subjects with mild cognitive impairment. Dement Geriatr Cogn Disord 2013;35:325-36.
141. Barten DM, Fanara P, Andorfer C, et al. Hyperdynamic microtubules, cognitive deficits, and pathology are improved in tau transgenic mice with low doses of the microtubule-stabilizing agent BMS-241027. J Neurosci 2012;32:7137-45.
142. Brunden KR, Zhang B, Carroll J, et al. Epothilone D improves microtubule density, axonal integrity, and cognition in a transgenic mouse model of tauopathy. J Neurosci 2010;30:13861-6.
143. Clavaguera F, Bolmont T, Crowther RA, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol 2009;11:909-13.
144. Boluda S, Iba M, Zhang B, Raible KM, Lee VM, Trojanowski JQ. Differential induction and spread of tau pathology in young PS19 tau transgenic mice following intracerebral injections of pathological tau from Alzheimer’s disease or corticobasal degeneration brains. Acta Neuropathol 2015;129:221-37.
145. Gibbons GS, Lee VMY, Trojanowski JQ. Mechanisms of cell-to-cell transmission of pathological Tau: a review. JAMA Neurol 2019;76:101-8.
146. Wang Y, Balaji V, Kaniyappan S, et al. The release and trans-synaptic transmission of Tau via exosomes. Mol Neurodegener 2017;12:5.
147. Pérez M, Avila J, Hernández F. Propagation of Tau via extracellular vesicles. Front Neurosci 2019;13:698.
150. Kuang H, Tan CY, Tian HZ, et al. Exploring the bi-directional relationship between autophagy and Alzheimer’s disease. CNS Neurosci Ther 2020;26:155-66.
151. Meco A, Li JG, Blass BE, Abou-Gharbia M, Lauretti E, Praticò D. 12/15-lipoxygenase inhibition reverses cognitive impairment, brain amyloidosis, and Tau pathology by stimulating autophagy in aged triple transgenic mice. Biol Psychiatry 2017;81:92-100.
152. Silva MC, Nandi GA, Tentarelli S, et al. Prolonged tau clearance and stress vulnerability rescue by pharmacological activation of autophagy in tauopathy neurons. Nat Commun 2020;11:3258.
153. Zhang ZH, Wu QY, Zheng R, et al. Selenomethionine mitigates cognitive decline by targeting both Tau hyperphosphorylation and autophagic clearance in an Alzheimer’s disease mouse model. J Neurosci 2017;37:2449-62.
154. Tang M, Ji C, Pallo S, Rahman I, Johnson GVW. Nrf2 mediates the expression of BAG3 and autophagy cargo adaptor proteins and tau clearance in an age-dependent manner. Neurobiol Aging 2018;63:128-39.
155. Smith PY, Hernandez-Rapp J, Jolivette F, et al. miR-132/212 deficiency impairs tau metabolism and promotes pathological aggregation in vivo. Hum Mol Genet 2015;24:6721-35.
156. Shibuya Y, Niu Z, Bryleva EY, et al. Acyl-coenzyme A:cholesterol acyltransferase 1 blockage enhances autophagy in the neurons of triple transgenic Alzheimer’s disease mouse and reduces human P301L-tau content at the presymptomatic stage. Neurobiol Aging 2015;36:2248-59.
157. Sun H, Zhong Y, Zhu X, et al. A tauopathy-homing and autophagy-activating nanoassembly for specific clearance of pathogenic Tau in Alzheimer’s disease. ACS Nano 2021;15:5263-75.
158. Tung YT, Wang BJ, Hu MK, et al. Autophagy: a double-edged sword in Alzheimer’s disease. J Biosci 2012;37:157-65.
159. Sandusky-Beltran LA, Sigurdsson EM. Tau immunotherapies: Lessons learned, current status and future considerations. Neuropharmacology 2020;175:108104.
160. Bittar A, Bhatt N, Kayed R. Advances and considerations in AD tau-targeted immunotherapy. Neurobiol Dis 2020;134:104707.
161. Rosenmann H, Grigoriadis N, Karussis D, et al. Tauopathy-like abnormalities and neurologic deficits in mice immunized with neuronal tau protein. Arch Neurol 2006;63:1459-67.
162. Kontsekova E, Zilka N, Kovacech B, Novak P, Novak M. First-in-man tau vaccine targeting structural determinants essential for pathological tau-tau interaction reduces tau oligomerisation and neurofibrillary degeneration in an Alzheimer’s disease model. Alzheimers Res Ther 2014;6:44.
163. Novak P, Schmidt R, Kontsekova E, et al. Safety and immunogenicity of the tau vaccine AADvac1 in patients with Alzheimer’s disease: a randomised, double-blind, placebo-controlled, phase 1 trial. Lancet Neurol 2017;16:123-34.
164. Novak P, Schmidt R, Kontsekova E, et al. FUNDAMANT: an interventional 72-week phase 1 follow-up study of AADvac1, an active immunotherapy against tau protein pathology in Alzheimer’s disease. Alzheimers Res Ther 2018;10:108.
165. Novak P, Zilka N, Zilkova M, et al. AADvac1, an active immunotherapy for Alzheimer’s disease and non Alzheimer tauopathies: an overview of preclinical and clinical development. J Prev Alzheimers Dis 2019;6:63-9.
166. Novak P, Kovacech B, Katina S, et al. ADAMANT: a placebo-controlled randomized phase 2 study of AADvac1, an active immunotherapy against pathological tau in Alzheimer’s disease. Nat Aging 2021;1:521-34.
167. Theunis C, Crespo-Biel N, Gafner V, et al. Efficacy and safety of a liposome-based vaccine against protein Tau, assessed in tau.P301L mice that model tauopathy. PLoS One 2013;8:e72301.
168. Rozenstein-Tsalkovich L, Grigoriadis N, Lourbopoulos A, et al. Repeated immunization of mice with phosphorylated-tau peptides causes neuroinflammation. Exp Neurol 2013;248:451-6.
169. Boutajangout A, Ingadottir J, Davies P, Sigurdsson EM. Passive immunization targeting pathological phospho-tau protein in a mouse model reduces functional decline and clears tau aggregates from the brain. J Neurochem 2011;118:658-67.
170. Dai CL, Chen X, Kazim SF, et al. Passive immunization targeting the N-terminal projection domain of tau decreases tau pathology and improves cognition in a transgenic mouse model of Alzheimer disease and tauopathies. J Neural Transm (Vienna) 2015;122:607-17.
171. Buée L, Bussière T, Buée-scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev 2000;33:95-130.
172. Collin L, Bohrmann B, Göpfert U, Oroszlan-Szovik K, Ozmen L, Grüninger F. Neuronal uptake of tau/pS422 antibody and reduced progression of tau pathology in a mouse model of Alzheimer’s disease. Brain 2014;137:2834-46.
173. Troquier L, Caillierez R, Burnouf S, et al. Targeting phospho-Ser422 by active Tau Immunotherapy in the THYTau22 mouse model: a suitable therapeutic approach. Curr Alzheimer Res 2012;9:397-405.
174. Sopko R, Golonzhka O, Arndt J, et al. Characterization of tau binding by gosuranemab. Neurobiol Dis 2020;146:105120.
175. Boxer AL, Qureshi I, Ahlijanian M, et al. Safety of the tau-directed monoclonal antibody BIIB092 in progressive supranuclear palsy: a randomised, placebo-controlled, multiple ascending dose phase 1b trial. Lancet Neurol 2019;18:549-58.
176. Bright J, Hussain S, Dang V, et al. Human secreted tau increases amyloid-beta production. Neurobiol Aging 2015;36:693-709.
177. Folch J, Petrov D, Ettcheto M, et al. Current research therapeutic strategies for Alzheimer’s disease treatment. Neural Plast 2016;2016:8501693.
178. Yanamandra K, Patel TK, Jiang H, et al. Anti-tau antibody administration increases plasma tau in transgenic mice and patients with tauopathy. Sci Transl Med 2017;9:eaal2029.
179. Qureshi IA, Tirucherai G, Ahlijanian MK, Kolaitis G, Bechtold C, Grundman M. A randomized, single ascending dose study of intravenous BIIB092 in healthy participants. Alzheimers Dement (N Y) 2018;4:746-55.
180. Lee SH, Le Pichon CE, Adolfsson O, et al. Antibody-mediated targeting of Tau In vivo does not require effector function and microglial engagement. Cell Rep 2016;16:1690-700.
181. .
182. Chai X, Wu S, Murray TK, et al. Passive immunization with anti-Tau antibodies in two transgenic models: reduction of Tau pathology and delay of disease progression. J Biol Chem 2011;286:34457-67.
183. Jicha GA, Bowser R, Kazam IG, Davies P. Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res 1997;48:128-32.
184. Luo W, Liu W, Hu X, Hanna M, Caravaca A, Paul SM. Microglial internalization and degradation of pathological tau is enhanced by an anti-tau monoclonal antibody. Sci Rep 2015;5:11161.
185. Vitale F, Giliberto L, Ruiz S, Steslow K, Marambaud P, d’Abramo C. Anti-tau conformational scFv MC1 antibody efficiently reduces pathological tau species in adult JNPL3 mice. Acta Neuropathol Commun 2018;6:82.
186. Sigurdsson EM. Tau immunotherapies for Alzheimer’s disease and related tauopathies: progress and potential pitfalls. J Alzheimers Dis 2018;64:S555-65.
187. Galpern WR, Mercken M, Van Kolen K, et al. P1-052: a single ascending dose study to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of the anti-phospho-tau antibody jnj-63733657 in healthy subjects. Alzheimers Dement 2019;15:P252-3.
188. A study to investigate safety and tolerability, pharmacokinetics and pharmacodynamics of JNJ-63733657 in healthy subjects and subjects with Alzheimer’s disease. Available from: https://clinicaltrials.gov [Last accessed on 20 Jul 2022].
189. Albert M, Mairet-Coello G, Danis C, et al. Prevention of tau seeding and propagation by immunotherapy with a central tau epitope antibody. Brain 2019;142:1736-50.
190. Courade JP, Angers R, Mairet-Coello G, et al. Epitope determines efficacy of therapeutic anti-Tau antibodies in a functional assay with human Alzheimer Tau. Acta Neuropathol 2018;136:729-45.
191. Kondo A, Shahpasand K, Mannix R, et al. Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature 2015;523:431-6.
192. Albayram O, Kondo A, Mannix R, et al. Cis P-tau is induced in clinical and preclinical brain injury and contributes to post-injury sequelae. Nat Commun 2017;8:1000.
193. Mohsenian Sisakht A, Karamzade-Ziarati N, Jahanbakhshi A, et al. Pathogenic cis p-tau levels in CSF reflects severity of traumatic brain injury. Neurol Res 2022;44:496-502.
194. Qiu C, Albayram O, Kondo A, et al. Cis P-tau underlies vascular contribution to cognitive impairment and dementia and can be effectively targeted by immunotherapy in mice. Sci Transl Med 2021;13:eaaz7615.
195. Nobuhara CK, DeVos SL, Commins C, et al. Tau antibody targeting pathological species blocks neuronal uptake and interneuron propagation of Tau in vitro. Am J Pathol 2017;187:1399-412.
196. Czerkowicz J, Chen W, Wang Q, et al. PAN-TAU ANTIBODY BIIB076 EXHIBITS PROMISING SAFETY AND BIOMARKER PROFILE IN CYNOMOLGUS MONKEY TOXICITY STUDY. Alzheimers Dement 2017;13:P1271.
197. Funk KE, Mirbaha H, Jiang H, Holtzman DM, Diamond MI. Distinct therapeutic mechanisms of Tau antibodies: promoting microglial clearance versus blocking neuronal uptake. J Biol Chem 2015;290:21652-62.
198. Kfoury N, Holmes BB, Jiang H, Holtzman DM, Diamond MI. Trans-cellular propagation of Tau aggregation by fibrillar species. J Biol Chem 2012;287:19440-51.
199. West T, Hu Y, Verghese PB, et al. Preclinical and clinical development of ABBV-8E12, a humanized anti-Tau antibody, for treatment of Alzheimer’s disease and other tauopathies. J Prev Alzheimers Dis 2017;4:236-41.
200. Höglinger GU, Litvan I, Mendonca N, et al. Safety and efficacy of tilavonemab in progressive supranuclear palsy: a phase 2, randomised, placebo-controlled trial. Lancet Neurol 2021;20:182-92.
201. Roberts M, Sevastou I, Imaizumi Y, et al. Pre-clinical characterisation of E2814, a high-affinity antibody targeting the microtubule-binding repeat domain of tau for passive immunotherapy in Alzheimer’s disease. Acta Neuropathol Commun 2020;8:13.
202. Rosenqvist N, Asuni AA, Andersson CR, et al. Highly specific and selective anti-pS396-tau antibody C10.2 targets seeding-competent tau. Alzheimers Dement (N Y) 2018;4:521-34.
203. Andersson CR, Falsig J, Stavenhagen JB, et al. Antibody-mediated clearance of tau in primary mouse microglial cultures requires Fcγ-receptor binding and functional lysosomes. Sci Rep 2019;9:4658.
204. Gao C, Chu X, Gong W, et al. Neuron tau-targeting biomimetic nanoparticles for curcumin delivery to delay progression of Alzheimer’s disease. J Nanobiotechnology 2020;18:71.
205. Yang Z, Liu ZW, Allaker RP, et al. A review of nanoparticle functionality and toxicity on the central nervous system. J R Soc Interface 2010;7 Suppl 4:S411-22.
206. Nguyen TT, Dung Nguyen TT, Vo TK, et al. Nanotechnology-based drug delivery for central nervous system disorders. Biomed Pharmacother 2021;143:112117.
207. Ghalandari B, Asadollahi K, Shakerizadeh A, et al. Microtubule network as a potential candidate for targeting by gold nanoparticle-assisted photothermal therapy. J Photochem Photobiol B 2019;192:131-40.
208. Sonawane SK, Ahmad A, Chinnathambi S. Protein-capped metal nanoparticles inhibit Tau aggregation in Alzheimer’s disease. ACS Omega 2019;4:12833-40.
209. Bajracharya R, Caruso AC, Vella LJ, Nisbet RM. Current and emerging strategies for enhancing antibody delivery to the brain. Pharmaceutics 2021;13:2014.
210. Wolfram J, Zhu M, Yang Y, et al. Safety of nanoparticles in medicine. Curr Drug Targets 2015;16:1671-81.
211. Liang Y, Duan L, Lu J, Xia J. Engineering exosomes for targeted drug delivery. Theranostics 2021;11:3183-95.
212. Qi Y, Guo L, Jiang Y, Shi Y, Sui H, Zhao L. Brain delivery of quercetin-loaded exosomes improved cognitive function in AD mice by inhibiting phosphorylated tau-mediated neurofibrillary tangles. Drug Deliv 2020;27:745-55.
213. Congdon EE, Chukwu JE, Shamir DB, et al. Tau antibody chimerization alters its charge and binding, thereby reducing its cellular uptake and efficacy. EBioMedicine 2019;42:157-73.
214. Congdon EE, Lin Y, Rajamohamedsait HB, et al. Affinity of Tau antibodies for solubilized pathological Tau species but not their immunogen or insoluble Tau aggregates predicts in vivo and ex vivo efficacy. Mol Neurodegener 2016;11:62.
215. Shamir DB, Deng Y, Wu Q, Modak S, Congdon EE, Sigurdsson EM. Dynamics of internalization and intracellular interaction of Tau antibodies and human pathological Tau protein in a human neuron-like model. Front Neurol 2020;11:602292.
216. Gu J, Congdon EE, Sigurdsson EM. Two novel Tau antibodies targeting the 396/404 region are primarily taken up by neurons and reduce Tau protein pathology. J Biol Chem 2013;288:33081-95.
217. Castillo-Carranza DL, Sengupta U, Guerrero-Muñoz MJ, et al. Passive immunization with Tau oligomer monoclonal antibody reverses tauopathy phenotypes without affecting hyperphosphorylated neurofibrillary tangles. J Neurosci 2014;34:4260-72.
218. Congdon EE, Gu J, Sait HB, Sigurdsson EM. Antibody uptake into neurons occurs primarily via clathrin-dependent Fcγ receptor endocytosis and is a prerequisite for acute tau protein clearance. J Biol Chem 2013;288:35452-65.
219. Tashima T. Delivery of intravenously administered antibodies targeting Alzheimer’s disease-relevant Tau species into the brain based on receptor-mediated transcytosis. Pharmaceutics 2022;14:411.
220. McEwan WA, Falcon B, Vaysburd M, et al. Cytosolic Fc receptor TRIM21 inhibits seeded tau aggregation. Proc Natl Acad Sci U S A 2017;114:574-9.
221. Bergen M, Barghorn S, Biernat J, Mandelkow EM, Mandelkow E. Tau aggregation is driven by a transition from random coil to beta sheet structure. Biochim Biophys Acta 2005;1739:158-66.
222. Rai SK, Savastano A, Singh P, Mukhopadhyay S, Zweckstetter M. Liquid-liquid phase separation of tau: from molecular biophysics to physiology and disease. Protein Sci 2021;30:1294-314.
223. Ambadipudi S, Biernat J, Riedel D, Mandelkow E, Zweckstetter M. Liquid-liquid phase separation of the microtubule-binding repeats of the Alzheimer-related protein Tau. Nat Commun 2017;8:275.
224. Strittmatter WJ, Saunders AM, Goedert M, et al. Isoform-specific interactions of apolipoprotein E with microtubule-associated protein tau: implications for Alzheimer disease. Proc Natl Acad Sci U S A 1994;91:11183-6.
225. Wang C, Xiong M, Gratuze M, et al. Selective removal of astrocytic APOE4 strongly protects against tau-mediated neurodegeneration and decreases synaptic phagocytosis by microglia. Neuron 2021;109:1657-1674.e7.
226. Shi Y, Manis M, Long J, et al. Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J Exp Med 2019;216:2546-61.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Guo Y, Li S, Zeng LH, Tan J. Tau-targeting therapy in Alzheimer's disease: critical advances and future opportunities. Ageing Neur Dis 2022;2:11. http://dx.doi.org/10.20517/and.2022.16
AMA Style
Guo Y, Li S, Zeng LH, Tan J. Tau-targeting therapy in Alzheimer's disease: critical advances and future opportunities. Ageing and Neurodegenerative Diseases. 2022; 2(3): 11. http://dx.doi.org/10.20517/and.2022.16
Chicago/Turabian Style
Guo, Yi, Song Li, Ling-Hui Zeng, Jun Tan. 2022. "Tau-targeting therapy in Alzheimer's disease: critical advances and future opportunities" Ageing and Neurodegenerative Diseases. 2, no.3: 11. http://dx.doi.org/10.20517/and.2022.16
ACS Style
Guo, Y.; Li S.; Zeng L.H.; Tan J. Tau-targeting therapy in Alzheimer's disease: critical advances and future opportunities. Ageing. Neur. Dis. 2022, 2, 11. http://dx.doi.org/10.20517/and.2022.16
About This Article
Copyright

Data & Comments
Data


 Cite This Article 113 clicks
Cite This Article 113 clicks

 Like This Article 40
likes
Like This Article 40
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.