
A sequential deposition of amyloid beta oligomers, plaques and phosphorylated tau occurs throughout life in the canine retina

Abstract
Aims: Cerebral amyloid burdens may be found in otherwise cognitively intact adults, often not showing worsening deficits with passing years. Alzheimer’s transgenic rodents have been widely used to investigate this phenomenon, but a spontaneous disorder in other animals, such as dogs that cohabit with humans and thus may have some shared environmental risks, may contribute and offer opportunities not possible in the smaller laboratory animals. In animals, the spontaneous disorder most comparable to Alzheimer’s disease (AD) affects mature to aged dogs and is designated canine cognitive dysfunction. Motivated by AD, many studies have revealed that amyloid progressively accumulates in the canine central nervous system, including the retina. Here, we investigated whether deposits of amyloid and/or tau can be found in the canine retina of neurologically normal animals from the first year of life to the elderly. Suppose canine ocular amyloid and tau are present from early life. In that case, that raises the question of whether similar patterns of accumulation occur in man, whether as part of aging, AD, or other.
Methods: This study used eye tissues from 30 dogs with a variety of ophthalmic or other orbital disorders, of which 7/30 were 1-2 years old. Tissues were subdivided into dogs of three different age groups: young (1-5 years old), middle (6-10 years old), and old (≥ 11 years old).
Results: Following immunostaining of tissue sections with nanobodies against retinal Aβ1-40 and Aβ1-42 oligomers, and antibodies against Aβ plaques (Aβp) and hyperphosphorylated Tau (p-Tau), our investigations revealed that accumulation of Aβ1-40 and Aβ1-42 oligomers were widespread in the retina in all age groups. In contrast, Aβp were detected in the middle and old age groups but not in the young age group. Furthermore, p-Tau staining was observed in four old dogs only, while other dogs were p-Tau free. Interestingly, both Aβo and Aβp co-localized in the middle and old age groups of dogs. Moreover, diffuse granular p-Tau co-localized with intracellular Aβo in the old age group. Finally, we also observed co-localization of Aβo and Aβp in the retinal vasculature which might be similar to cerebral amyloid angiopathy associated with AD.
Conclusion: As far as we know, the presence of amyloid and tau in the canine retina is hitherto unreported. If similar, early-in-life subclinical retinal deposits occur in a human cohort perhaps identified by AD genetic risk factors, following this group may offer the prospect of preclinical therapeutic intervention in imminent dementia, a strategy recognized as likely necessary to impact this burgeoning disorder.
Keywords
INTRODUCTION
The principal neuropathological lesions in Alzheimer’s disease (AD) brains include extracellular neuritic and/or diffuse plaques containing amyloid-beta (Aβ), intracellular tau protein (p-Tau) in the form of neurofibrillary tangles (NFTs) in addition to cerebral amyloid angiopathy (CAA), ubiquitin, severe synaptic loss, neuronal death, and brain atrophy[1-4]. While the deposition of Aβ in the human brain has traditionally been accepted as a major hallmark of AD[5], its accumulation is also observed in about 20% of cognitively unimpaired, aged individuals[6,7]. For decades, the association of Aβ with AD has been demonstrated, but the significance and impact of Aβ accumulation in healthy individuals are both poorly understood[8] and a source of confusion. For example, previous studies did not establish a clear correlation with memory loss in the aging brain[9-11]. Previous reports documented the progressive accumulation of both Aβ plaques (Aβp) in the brain[12,13] and Aβ oligomers (Aβo) in the brain and periphery of cognitively unimpaired individuals[14,15]. A study by Lesne et al. measured the levels of three Aβo “species”, including Aβ trimers, Aβ*56 and Aβ dimers, in brain tissues from 75 cognitively unimpaired individuals, including young children and adolescents[14]. The authors showed that Aβ trimers were present in the central nervous system (CNS) of children and adolescents, and their levels increased progressively with age, suggesting that this particular
Currently available transgenic animal models of AD do not replicate the subtle clinical and pathological features of the disease, as demonstrated by their lack of reproducible therapeutic outcomes[18,19]. As to the dog’s age, there is an anatomically defined accumulation of Aβ peptides in the CNS, while less is known about cerebral p-Tau deposition. Some animals will develop a progressive cognitive decline with loss of learned behaviors and memory, a syndrome named canine cognitive dysfunction (CCD)[20-24], with CNS changes similar to the neuropathological hallmarks characteristic of AD[25-28]. In those destined to develop AD, amyloid precursor protein (APP) is sequentially cleaved by β and γ secretase leading to the production of Aβ peptide fragments (36-43 amino acids), which then aggregate and deposit as plaques[4,5,29]. The canine APP and Aβ peptides are 98% and 100% identical to their human counterparts[24]. In older dogs and others affected with CCD, Aβ deposits as diffuse plaques, while the dense core of humans’ mature plaques is very rare or absent[20,21,27,30].
There are two major isoforms of Aβ, Aβ1-40 (~80%-90%) and Aβ1-42 (~5%-10%) and three major assemblies[31-34] recognized in man and animals/dogs, including monomeric Aβ, Aβo containing 12-24 monomers which become elongated to form protofibrils and finally insoluble fibrils[35]. Among these three stages, Aβo is thought to be the most toxic to neurons and responsible for the structural and functional pathologic changes associated with AD[36-39]. Likewise, a previous study reported that cognitive decline occurs before the accumulation of Aβp in CCD, supporting the premise that earlier assembly states of Aβ may be the toxic species[23]. In addition, CAA, ubiquitin, and severe synaptic loss have also been reported in CCD[20,40-43]. Another cardinal pathological hallmark of AD is the accumulation of p-Tau[3,4]. Several phosphorylation sites were identified on tau in aged dogs including Thr181[42], Ser422[43], Ser202/Thr205[40,43], Ser396[30,40,43], Ser189, and Ser207[44]; however, demonstrating the presence of NFTs has been infrequent compared to the consistent demonstration of amyloid oligomers and diffuse plaques. A study by Schmidt and colleagues using anti-pT205, AT8, AT100, PHF-1, and anti-pT422 antibodies seeking the presence of tau pathology in 24 dogs aged between two and nineteen years, showed that three 13-15-year-old dogs displayed p-Tau and only one 15-year-old Pekingese dog displayed NFT-like appearance[43].
In AD, visual disturbances are one of the early complaints and include loss of color vision, impairment of peripheral vision and object recognition, contrast sensitivity, and decreased visual memory and perception[45-47]. A recent longitudinal study of 1349 older adults showed that poor visual acuity paralleled the development of dementia[48], suggesting the possibility that ocular disturbances can be used as an early predictor of dementia risk in the older population[48]. Moreover, post-mortem studies of AD and animal models of AD demonstrated a strong association between retinal accumulation of Aβo[49,50]. Aβ plaques[51,52], p-Tau[53,54] and brain depositions and cognitive decline, where retinal Aβo was shown to deposit earlier than the brain and before deficits in cognition. Although, to our knowledge, age-dependent retinal deposition of Aβo and/or Aβp has not been investigated in AD and cognitively unimpaired individuals, including children and adolescents, our recent studies in AD mice models confirmed the conversion of cerebral and retinal Aβo to Aβp in an age-dependent manner, where retinal Aβo was detected as early as 3-month old APP/PS1 mice, before brain pathology and cognitive decline were observed[49,55].
In this study, we investigated the retinal accumulation of Aβo, Aβp, and p-Tau in three age groups of genetically diverse and neurologically intact populations of dogs. Here, following immunohistochemistry and immunofluorescence analysis, we confirmed the presence of retinal Aβ40 and Aβ42 oligomers, Aβp, and p-Tau. Retinal Aβ40 and Aβ42 oligomers deposition was conspicuous and widespread and observed in all age groups, including the young 1-5-year-old neurologically intact group. Moreover, retinal co-localization of Aβ40/Aβ42 oligomers and Aβp was observed in few middle-aged dogs and most dogs in the old neurologically intact age group, while retinal co-localization of Aβ40/Aβ42 oligomers and p-Tau was only seen in the old neurologically intact age group of dogs. Morphologically, extracellular Aβp deposits appeared as small, dot-like rounded deposits, while intracellular p-Tau deposits adopted a diffuse appearance in the retinal layers[54]. No NFTs or neuropil threads were observed in these dogs. Taken together, these results highlight the importance of further investigations of AD-related pathology in the retina of presymptomatic children and adolescents to gain insight into disease progression and potentially identify early at-risk individuals to help implement speedy therapeutic interventions.
METHODS
Dog eye samples and animal ethics
Sections of eyes used in this study were prepared from archived cases in the Comparative Ocular Pathology Laboratory of Wisconsin (COPLOW) at the Department of Pathobiological Sciences, School of Veterinary Medicine, University of Wisconsin, Madison. Surgical enucleations had been submitted by practicing veterinarians to COPLOW for routine pathological diagnosis. These canine eyes came from 30 random, genetically diverse dogs (15 different breeds) ranging from 1 to 16 years of age with a broad variety of ocular disorders [Tables 1 and 2]. Such tissues, historically submitted for disease investigation purposes, are not subject to approval by institutional animal ethical regulations.
Signalment for 30 dogs and their retinal Aβ oligomers and plaques IHC scores
| Age groups | Age (year) | Sex | Breed | Size | Pathological Aβ IHC findings in the dog retina | |
| A11 - Aβ oligomers | 4G8 - Aβ plaques | |||||
| Young (1-5 years) | 1 | SF | Staffordshire terrier | Medium | + | - |
| 1.4 | M | Siberian husky | Medium | - | - | |
| 1.8 | SF | Mixed breed | ND* | +++ | - | |
| 1.10 | SF | Siberian husky | Medium | ++ | - | |
| 2 | F | Chihuahua mix | Toy | +++ | - | |
| 2 | M | Bichon frise mix | Small | + | - | |
| 2 | NM | Shih Tzu | Small | + | - | |
| 3 | NM | German shepherd | Large | +++ | - | |
| 3 | F | Giant schnauzer | Large | + | - | |
| 3.6 | M | Siberian husky | Medium | + | - | |
| Middle age (6-10 years) | 7 | NM | Hound mixed | Medium | - | - |
| 8 | SF | Bedlington terrier | Small | - | - | |
| 8 | SF | Cocker spaniel | Medium | - | - | |
| 8 | M | Mixed breed | ND* | - | - | |
| 8 | M | Cocker spaniel | Medium | + | - | |
| 8 | F | German shepherd mix | Large | - | - | |
| 8 | SF | Great dane dog | Giant | - | - | |
| 9 | M | Jack Russell terrier dog | Small | - | - | |
| 9.5 | SF | Boxer dog | Large | - | - | |
| 9.9 | SF | Cocker spaniel | Medium | - | - | |
| Elderly (11-16 years) | 11 | NM | Mixed breed | ND* | - | - |
| 11 | NM | Beagle | Small | - | - | |
| 11.9 | SF | Bouvier des flanders | Large | + | - | |
| 12 | SF | German shepherd | Large | - | ++ | |
| 12 | SF | German shepherd mix | Large | - | - | |
| 12 | NM | Husky mix | Medium | - | - | |
| 12.8 | NM | Shih-tzu | Small | - | ++ | |
| 13 | NM | Border collie | Medium | - | - | |
| 15 | SF | Basset hound | Medium | - | + | |
| 16 | SF | Shih Tzu | Small | - | - | |
Signalment for 30 dogs, their ophthalmological disorders, and retinal Aβ and p-Tau IF scores
| Age groups | Age (year) | Sex | Breed | Size | Ophthalmological disorders which resulted in enucleation | Pathological IF findings in the dog retina | |||
| Aβ40 oligomers | Aβ42 oligomers | p-Tau | |||||||
| Young (1-5 years) | 1 | SF | Staffordshire terrier | Medium | Anterior segment dysgenesis | + | + | - | - |
| 1.4 | M | Siberian husky | Medium | Anterior segment dysgenesis | - | - | - | - | |
| 1.8 | SF | Mixed breed | ND* | Anterior segment dysgenesis | +++ | +++ | - | - | |
| 1.10 | SF | Siberian husky | Medium | Anterior segment dysgenesis | +++ | +++ | - | - | |
| 2 | F | Chihuahua mix | Toy | Anterior segment dysgenesis | +++ | +++ | - | - | |
| 2 | M | Bichon frise mix | Small | Proptosis | +++ | +++ | - | - | |
| 2 | NM | Shih Tzu | Small | Melanoma limbal | ++ | ++ | - | - | |
| 3 | NM | German shepherd | Large | Rhabdomyosarcoma orbital | +++ | +++ | - | - | |
| 3 | F | Giant schnauzer | Large | Phthisis bulbi | ++ | ++ | - | - | |
| 3.6 | M | Siberian husky | Medium | Anterior segment dysgenesis | ++ | ++ | - | - | |
| Middle age (6-10 years) | 7 | NM | Hound mixed | Medium | Hemangiosarcoma | - | - | - | - |
| 8 | SF | Bedlington terrier | Small | Neoplasia | - | - | - | - | |
| 8 | SF | Cocker spaniel | Medium | Conjunctivitis | +++ | +++ | + | - | |
| 8 | M | Mixed breed | ND* | Conjunctival melanoma | +++ | +++ | + | - | |
| 8 | M | Cocker spaniel | Medium | Pre glaucoma | +++ | +++ | +++ | - | |
| 8 | F | German shepherd mix | Large | Mast cell tumor | ++ | ++ | + | - | |
| 8 | SF | Great dane dog | Giant | Complex apocrine adenocarcinoma | + | + | - | - | |
| 9 | M | Jack Russell terrier dog | Small | Sialoadenitis | ++ | ++ | - | - | |
| 9.5 | SF | Boxer dog | Large | Hemangioma | + | + | - | - | |
| 9.9 | SF | Cocker spaniel | Medium | Adenocarcinoma a orbital | +++ | +++ | +++ | - | |
| Elderly (11-16 years) | 11 | NM | Mixed breed | ND* | Squamous cell carcinoma | - | - | - | - |
| 11 | NM | Beagle | Small | Melanoma conjunctival | +++ | +++ | +++ | - | |
| 11.9 | SF | Bouvier des flanders | Large | Melanoma eyelid | + | + | - | - | |
| 12 | SF | German shepherd | Large | Melanoma eyelid | +++ | +++ | +++ | +++ | |
| 12 | SF | German shepherd mix | Large | Melanoma conjunctival | +++ | +++ | +++ | - | |
| 12 | NM | Husky mix | Medium | Orbital carcinoma | +++ | +++ | ++ | +++ | |
| 12.8 | NM | Shih-tzu | Small | Glaucoma | +++ | +++ | ++ | +++ | |
| 13 | NM | Border collie | Medium | Melanoma skin | +++ | +++ | +++ | +++ | |
| 15 | SF | Basset hound | Medium | Conjunctival hemangiosarcoma | +++ | +++ | +++ | - | |
| 16 | SF | Shih Tzu | Small | Squamous cell carcinoma | + | + | - | - | |
Tissue preparation
Enucleated eyes were submitted to the laboratory fixed in 10% neutral buffered formalin. On receipt, they were examined for gross abnormalities before trimming, followed by processing overnight using a Leica ASP300S tissue processor (Leica biosystem, Wetzlar, Germany). Tissues were then embedded in paraffin blocks and sectioned at 4μm thickness using a Leica RM2235 microtome (Leica biosystem, Wetzlar, Germany) and placed on charged slides. Sections were then stained with hematoxylin and eosin (H&E) and Congo red (CR). Further sections were used for immunohistochemistry (IHC) and immunofluorescence (IF) staining, and both upper and lower parts of the retina were assessed.
Hematoxylin and eosin staining
Paraffin sections were dewaxed by two changes of absolute xylene for 5 min each. Sections were rehydrated using two changes of 100% ethanol for 2 min each, 95% ethanol for 3 min, 70% ethanol for 2 min, and finally rinsed in deionized water for 2 min. H&E stain was then performed by adding Gill ll Haematoxylin solution (Leica Biosystems, Wetzlar, Germany), followed by 1% acid alcohol and subsequently eosin stain (Leica Biosystems, Wetzlar, Germany). Finally, the sections were dehydrated with increasing concentrations of ethanol, from 70%, 95% to 100%, then dewaxed by two changes of xylene and finally mounted with xylene-based mounting media (Sigma Aldrich, Missouri, United States)[56]. H&E staining was used to assess retinal morphology, the neuronal population within the eye.
Congo red staining
Initially, the CR working solution was prepared by mixing 50 ml Congo red solution and 0.5 ml potassium hydroxide solution supplied in the Congo red amyloid special stain kit (Leica Biosystems, Wetzlar, Germany). Retinal sections were placed in the working solution for 20 min and then rinsed in 5-8 changes of deionized water. This was followed by staining with Gill ll Haematoxylin (Leica Biosystems, Wetzlar, Germany) for 1-3 min and rinsing in 3 changes of deionized water. Sections were then dehydrated in two changes of 95% alcohol followed by three changes of absolute alcohol for one minute each. Finally, the sections were cleared in two changes of xylene and mounted in a xylene miscible medium. Amyloid fibrils appeared as dull to red brick under light microscopy (Olympus CX 43, Shinjuku, Tokyo, Japan) and apple green birefringent under polarized light (Olympus CX 43, Shinjuku, Tokyo, Japan).
Immunohistochemical staining of amyloid-beta plaques and amyloid-beta oligomers
Eye sections that contained the retina were pre-treated using the 2100 antigen retriever (Aptum Biologics Ltd, Southampton, United Kingdom) to expose the target epitopes. The sections were then treated with 90% formic acid for 5 min at room temperature (RT) followed by cell membrane permeabilization, achieved by using 0.1% Triton X for 1 min before the addition of 0.3% H2O2 for 15 min to inactivate endogenous peroxidases. Sections were then blocked with protein block serum-free (Agilent, City, Country) for 15 min. The sections were then stained for 1 h with the following primary antibodies in phosphate-buffered saline (PBS): mouse purified 4G8 anti-Aβ17-24 (1:500; Bio legend, San Diego, CA, USA) or A11 rabbit anti-Aβo (1:250; Merck Millipore, Burlington, MA, USA) antibodies. After washing with PBS, sections were incubated for 1 h at RT with the following secondary antibodies in PBS: HRP-conjugated anti-mouse IgG (Sigma-Aldrich, Missouri, United States) or anti-rabbit IgG (Sigma-Aldrich, Missouri, United States). The sections were then washed three times with PBS before the addition of the 3,3'-Diaminobenzidine substrate chromogen system and incubated for 5-10 min. The sections were then counterstained with hematoxylin for 1 min before mounting. Sections were finally imaged using Olympus CX 43 light microscopy (Shinjuku, Tokyo, Japan).
The staining intensity was semi-quantitatively analyzed and scored across the retinal layers. Each bright field was examined at 40× magnification and categorized into no immunostaining “-”; low immunoreactivity, found in limited areas of the retinal layers “+”; moderate immunoreactivity where Aβ deposits were numerous “++”; and finally strong immunoreactivity with widespread A11 and 4G8 positive Aβ labeling “+++” [Table 1].
Immunofluorescence detection and co-localization studies
To investigate whether Aβo co-localized with Aβp or with p-Tau, we performed immunofluorescence double labeling using camelid-derived single domain anti-A40 (PRIOAD12) or anti-A42 (PRIOAD13) oligomer antibodies with 4G8 anti-Aβp antibody (Bio legend, San Diego, California, United States) or anti-phosphorylated tau AT8 antibody targeting amino acid residues Ser202/Thr205 (Thermo-fisher Scientific, Massachusetts, United States). Slides were processed as described above for IHC before adding the primary antibodies. The sections were double-stained with either PRIOAD 12 (1:500) or PRIOAD 13 (1:500)[57] and 4G8 antibody overnight (1:500) or AT8 (1:500) and either PRIOAD 12 (1:500) or PRIOAD 13 (1:500). Sections were then washed in Tris-buffered saline with 0.05% Tween 20 (TBST) and incubated with secondary antibodies at a dilution of 1:500, including goat anti-llama IgG conjugated to FITC (Bethyl Laboratories, Inc, Texas, USA) or donkey-anti-mouse IgG conjugated to Texas red (Sigma-Aldrich, Missouri, United States) for 2 hours at room temperature. Other sections were used as negative control and stained with secondary antibodies with the omission of the primary antibodies. Retinal sections derived from an APP/PS1 and TAU58/2 transgenic mice models[58,59] were used as positive control and stained with anti-Aβ and anti-P-Tau antibodies, respectively. The sections were covered, slipped with paramount aqueous mounting medium (Dako, Agilent, Santa Clara, California, United States), sealed, and dried overnight. Finally, the sections were visualized with LSM800 confocal microscope with a standard FITC/Texas Red double band-pass filter set (Zeiss, Oberkochen, Germany).
RESULTS
Histological assessment of the retina in 30 dogs
We performed H&E staining of the canine eye sections and examined the retinal layers to assess their general morphological appearance and identify pathological changes such as vacuolation, neuronal death, and eosinophilic deposits[49,55]. No specific lesions were observed in the retinal layers of the young and middle age groups (data not shown). However, scattered eosinophilic deposits in the ganglion cell layer (GCL) and inner nuclear layer (INL) of the retina were noticeable in 5/10 dogs in the older age group (11-16 years old), including an 11-year-old Beagle, a 12-year-old German shepherd, a 12-year-old Husky mix, a 12.8-year-old Shih Tzu, and a 15-year-old Basset hound [Figure 1A and B]. Furthermore, CR staining used to detect any amyloid fibrils and CAA in eye sections of the dogs did not display any staining in the retina tissues from all age groups (data not shown).
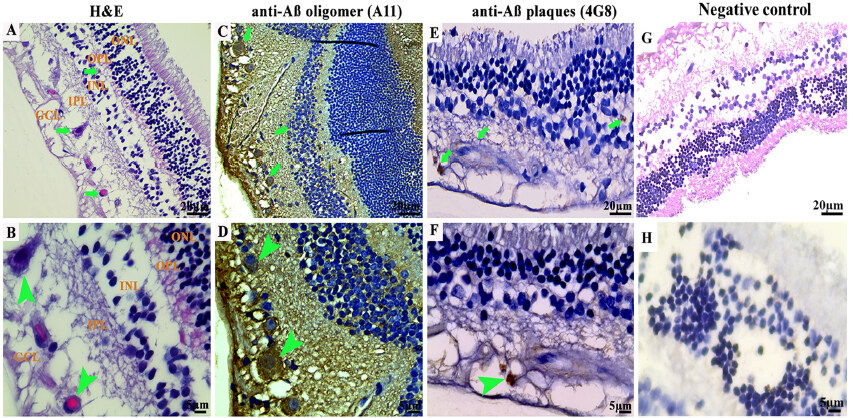
Figure 1. Photomicrographs of the microscopic lesions in the canine retina. The retina in a 12-year-old German shepherd dog. Eosinophilic deposits (green arrows) were observed in the ganglion cell layer (GCL) and inner nuclear layer (INL). Representative of 10 cases of old-aged dogs. Hematoxylin and eosin (H&E) 40×. (B) Higher magnification of the deposits in image (A) (green arrowhead) in the GCL. H&E 100×. (C) Retina of a 3-year-old German shepherd dog. Immunohistochemical staining with A11 anti-Aβo IgG antibody exhibited cytoplasmic Aβo depositions in the GCL and INL (green arrows). Representative of 10 cases of young age group. IHC 40×. (D) Higher magnification of deposits from image (C) in the GCL (green arrowheads). IHC 100×. (E) Retina of a 12-year-old German shepherd mix dog. Immunohistochemical staining with 4G8 anti-Aβp IgG antibody exhibited extracellular Aβ aggregates in the GCL and INL (green arrows). IHC 40×. (F) Higher magnification of deposit from image (E) in the GCL (green arrowhead). (G and H) Representative retinal sections derived from dogs of all age groups stained with secondary antibodies with the omission of the primary antibody did not show any depositions (40× and 100×, respectively). The photomicrographs were taken from the peripheral region of the retina - away from the optic disc. Representative of all 30 dogs examined. IHC 100×. IHC: Immunohistochemistry.
Immunohistochemical detection of Aβ oligomers and plaques in the retina of 30 dogs
While demonstrated in the CNS of aged animals including cats and bears[24,60], dogs have been more comprehensively studied and shown to develop human type Aβ deposition at a very young age. Retinal tissues stained with A11 exhibited intracellular Aβo depositions in the outer nuclear layer (ONL), INL, and GCL [Figure 1C and D]. The intraneuronal Aβo intensity following A11 staining ranged from low (+), moderate (++) to strong (+++) in all dogs in the young age group except a 1.4 years old male Siberian husky that did not display any A11 positive stain [Supplementary Figure 1A]. Of note, retinal sections derived from wild-type mice were used as negative control following immunofluorescence staining [Supplementary Figure 1B and C].
A spayed female mixed breed aged 1.8 years, a female Chihuahua aged 2 years, and a neutered male German shepherd aged 3 years showed strong (+++) accumulation of A11 positive Aβo [Figure 1C and D]. Two dogs displayed a low amount (+) of A11 positive Aβo in the retinal layers in the middle and old age groups, including an 8-year-old male Cocker spaniel and an 11.9-year-old spayed female Bouvier des Flanders [Table 1]. In addition, 4G8-positive small rounded and dot-like extracellular Aβp deposits were observed in the INL, inner plexiform layer (IPL), and GCL [Figure 1E and F] of the retina. They were structurally different from the typical large diffuse plaques normally observed in dog brains with CCD[21,30,61]. The extracellular Aβp intensity following 4G8 staining ranged from low (+) to moderate (++) in some dogs in the old age group, including a 15-year-old spayed female Basset hound, a 12.8-year-old neutered male Shih Tzu, and a 12-year-old spayed female German shepherd. Other dogs in this age group were all negative for 4G8 staining [Table 1, Figure 1E and F]. Representative retinal sections derived from dogs of all age groups stained with secondary antibodies with the omission of the primary antibody did not show any depositions [Figure 1G and H].
Immunofluorescence detection and co-localization of retinal Aβ40, Aβ42 oligomers, and Aβ plaques in the retina of 30 dogs
To confirm the presence of retinal Aβ40 and Aβ42 oligomers and to determine whether Aβ40 and/or Aβ42 oligomers co-localized with Aβp in the retinas of different age groups and breeds of neurologically intact dogs, we performed immunofluorescence double staining using PRIOAD12 (Aβ40 oligomers), PRIOAD13 (Aβ42 oligomers) camelid-derived single domain anti-Aβ oligomer and 4G8 anti-Aβp antibodies. Morphologically, Aβo appeared as globular and annular in shape[55,62] and deposited intracellularly in the ONL, INL, and GCL [Figures 2-4]. 4G8 positive Aβ plaques appeared morphologically as dot-like and small rounded extracellular deposits[50,54] in the outer plexiform layer (OPL), IPL, and GCL [Figures 2-4]. We found that both Aβ40 and Aβ42 oligomers staining was widespread in the majority of the dogs in young
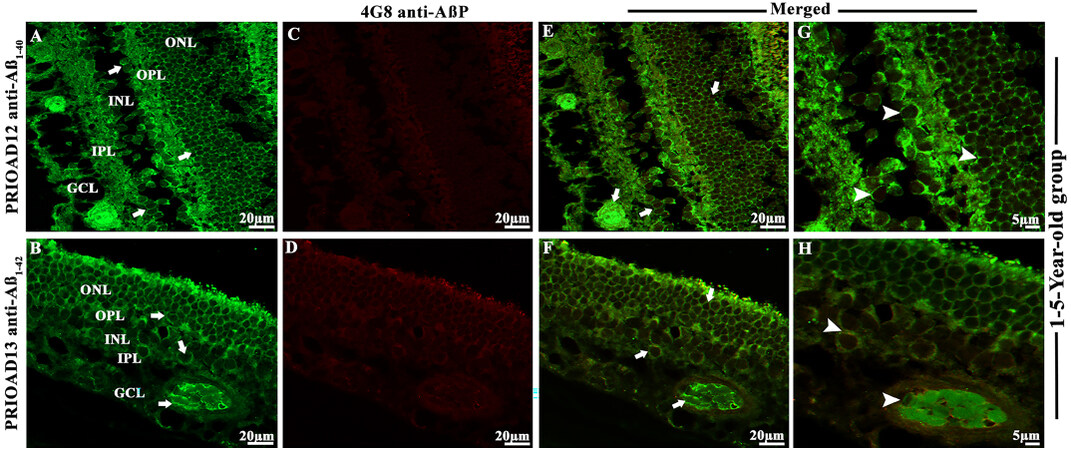
Figure 2. Immunofluorescence (IF) detection and co-localization of retinal amyloid-beta oligomers and amyloid-beta plaques in the dogs of the 1-5-year-old group. Retinal co-staining of oligomers with anti-Aβ40 (PRIOAD12) or anti-Aβ42 (PRIOAD 13) camelid-derived single domain antibodies (green) and plaques with anti-Aβ (4G8) antibody (red) of a 3-year-old German shepherd dog (A-H). A and B show widespread accumulation of Aβ40 and Aβ42 oligomers in the GCL, INL, and ONL (white arrows) and retinal vasculature, respectively. IF 40×. (C and D) No Aβp was detected in the retinal layers. IF 40×. (E and F) Co-localization of Aβo and Aβp was not observed in the retinal layers of this animal (Aβo was present - white arrows). G and H are higher magnification 100× of images (A and B) in the GCL and INL, ONL and retinal vasculature. Representative of 10 dogs in the younger age group (1-5 years). GCL: Ganglion cell layer; INL: inner nuclear layer; ONL: outer nuclear layer.
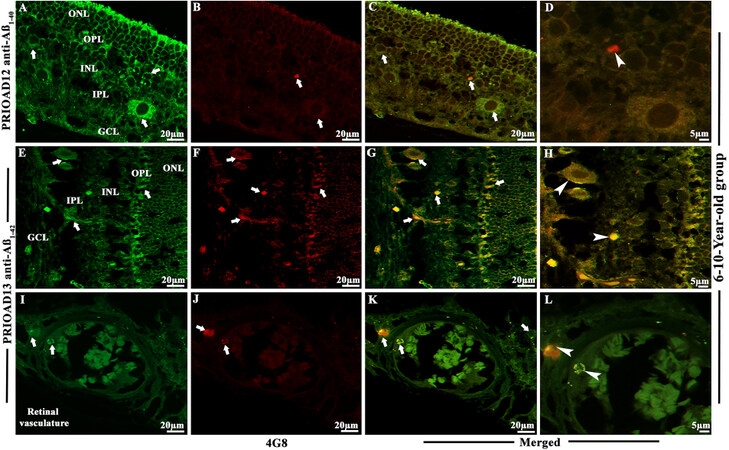
Figure 3. Immunofluorescence detection and co-localization of retinal amyloid-beta oligomers and amyloid-beta plaques in dogs of the 6-10-year-old group. Retinal co-staining with anti-Aβ40 (PRIOAD 12) and anti-Aβ42 (PRIOAD 13) camelid-derived single domain antibody (green) and 4G8 antibody (red) of a 9-year-old Cocker spaniel dog (A-L). (A) A large number of Aβ40 and (B) Aβ42 oligomers were found in the GCL, INL, and ONL (white arrows, 40×). (C) Detection of Aβ42 oligomers in the vasculature. 4G8 positive Aβ plaque-like deposits were observed in the (D and E) GCL, IPL, INL, and OPL of the retina (white arrows, 40×). Widespread co-localization was observed in the (G and H) retinal layers (white arrows, 40×). Co-localization of 4G8 positive Aβ plaque with (J) Aβ40 and (K) Aβ42 depositions (white arrowhead) showed with higher magnification (100×) in the GCL and INL of the same 9-year-old Cocker spaniel dog retinal section. (C) Aβ oligomers and (F) 4G8 positive Aβ plaques were observed in the retinal vasculature (white arrows). (I and L) Co-localization of Aβ oligomers and 4G8 positive Aβ plaques were exhibited with 40× and with higher 100× magnification in the retinal vessel wall, respectively (white arrowhead). The photomicrograph was derived from the peripheral region of the retina - away from the optic disc. Representative of 10 dogs examined from middle age group (6-10 years). GCL: Ganglion cell layer; IPL: inner plexiform layer; INL: inner nuclear layer; OPL: outer plexiform layer; ONL: outer nuclear layer.
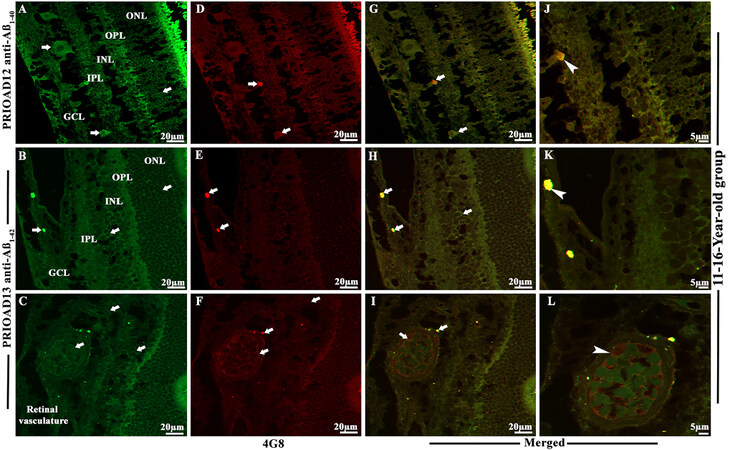
Figure 4. Immunofluorescence co-localization of retinal amyloid-beta oligomers and amyloid-beta plaques in dogs of 11-16-year-old group. Retinal co-staining with anti-Aβ40 (PRIOAD 12) and anti-Aβ42 (PRIOAD 13) camelid-derived single domain antibody (green) and 4G8 antibody (red) of a 12-year-old German shepherd dog (A-L). A large number of (A) Aβ40 and (B) Aβ42 oligomers were found in the GCL, INL, and ONL (white arrows, 40×). 4G8 positive Aβ plaque-like deposits were observed in the (D and E) GCL of the retina (white arrows, 40×). Widespread co-localization was observed in the (G and H) retinal layers (white arrows, 40×). Co-localization of 4G8 positive Aβ plaque with (J) Aβ40 and (K) Aβ42 depositions (white arrowhead) showed with higher magnification (100×) in the GCL of the same 12-year-old German shepherd dog retinal section. (C) Aβ oligomers and (F) 4G8 positive Aβ plaques were observed in the retinal vasculature (white arrows). (I and L) Co-localization of Aβ oligomers and 4G8 positive Aβ plaques were exhibited with 40× and also with higher 100× magnification in the retinal vessel wall, respectively (white arrowhead). The photomicrograph was derived from the peripheral region of the retina - away from the optic disc. Representative of 10 dogs examined from elder age group (11-16 years). GCL: ganglion cell layer; INL: inner nuclear layer; ONL: outer nuclear layer.
Immunofluorescence co-localization of retinal Aβ oligomers and phosphorylated tau in the retina of 30 dogs
To confirm the presence of p-Tau and to investigate whether p-Tau co-localizes with Aβo, we performed double fluorescence staining of Aβo and p-Tau using PRIOAD 12 (Aβ40) or PRIOAD 13 (Aβ42) anti-oligomer[57] and AT8 anti-p-Tau antibody, targeting Ser202/Thr205[54]. Aβo appeared as globular and annular in shape[55,62] [Figure 5A and B] and AT8 positive p-Tau appeared morphologically as diffuse and granular intracellular deposits[54] in the OPL, INL, IPL, and GCL [Figure 5C and D]. Overall, Aβo and p-Tau did not display consistent co-localization in all age groups, as P-tau was only detected in the old age group and Aβ40 or Aβ42 oligomers were identified in all age groups. Among ten dogs in the old age group, only four dogs have displayed the presence of p-Tau including a spayed female German shepherd and a neutered male Husky aged 12 years, a neutered male Shih Tzu aged 12.8 years, and a neutered male Border collie aged 13 years, and they exhibited strong co-localization with Aβ40 or Aβ42 oligomers [Figure 5E-H]. Representative retinal sections derived from Tau 58/2 mouse were used as positive control and confirmed the presence of AT8 positive p-Tau [Supplementary Figure 5D][58]. Representative retinal sections derived from dogs of all age groups stained with secondary antibodies with the omission of the primary antibody did not show any co-localization [Supplementary Figure 6A and B].
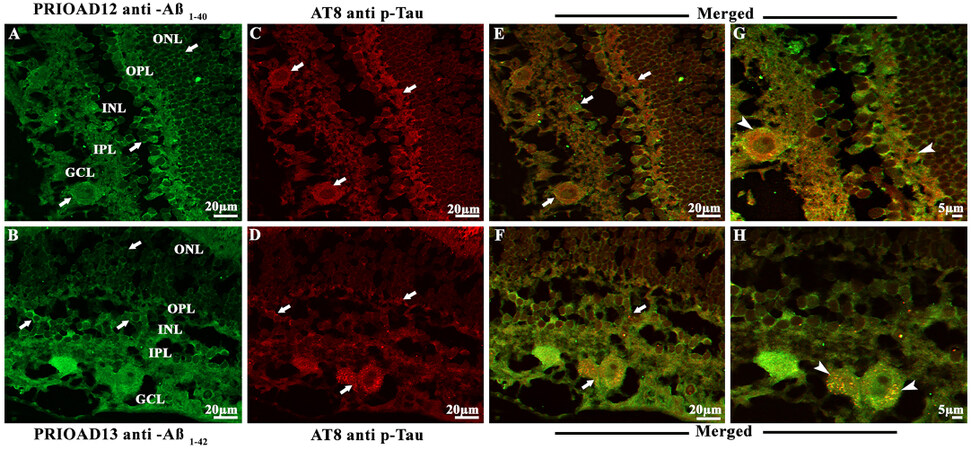
Figure 5. Immunofluorescence co-localization of retinal amyloid-beta oligomers and hyperphosphorylated tau in dogs of 11-16-year-old group. Retinal co-staining with anti-Aβ40 (PRIOAD 12) and anti-Aβ42 (PRIOAD 13) camelid-derived single domain antibody (green) and AT8 antibody (red) of a 12-year-old German shepherd dog. A large number of (A) Aβ40 and (B) Aβ42 oligomers were found in the GCL, INL, and ONL (white arrows, 40×). AT8 positive diffuse p-Tau-like deposits were observed in the (C and D) GCL, IPL, INL, and OPL of the retina (white arrows, 40×). Widespread co-localization was observed in the (E and F) retinal layers (white arrows, 40×). Co-localization of AT8 positive p-Tau with (G) Aβ40 and (H) Aβ42 depositions (white arrowhead) showed with higher magnification (100×) in the GCL and OPL of the same 12-year-old German shepherd dog retinal section. The photomicrograph was derived from the peripheral region of the retina - away from the optic disc. Representative of 10 dogs examined from elder age group (≥ 11 years). GCL: Ganglion cell layer; IPL: inner plexiform layer; INL: inner nuclear layer; OPL: outer plexiform layer; ONL: outer nuclear layer.
Influence of demographic factors on the retinal deposition of Aβ oligomers, Aβ plaques, and phosphorylated tau in 30 dogs
To investigate the influence of the demographic factors on retinal Aβ and p-Tau deposition, staining intensity was compared with the age, breed, size, and sex of the neurologically intact dogs [Table 2]. Aβ40 and Aβ42 oligomers, Aβp and p-Tau fluorescence intensity, were assessed at 40× magnification. Immunoreactivity throughout the retinal layers was semi-quantified and categorized into no immunostaining “-”; low immunoreactivity, exhibited in limited areas of the retinal layers “+”; moderate immunoreactivity where deposits were more apparent “++” and finally strong immunoreactivity with widespread Aβo, Aβp and p-Tau labeling “+++” [Table 2]. After comparing the age of the dogs and immunofluorescence staining scores, we found that both Aβ40 and Aβ42 oligomers staining was high in young dogs, which slightly decreased in the middle age group, then finally, an upward trend was noticed in older dogs [Table 2, Figure 6]. In comparison, Aβp was absent in young dogs, but its presence in the middle age group was moderate and high in the old age group [Table 2, Figure 6]. Finally, p-Tau deposits were not observed in the young and middle age groups, whereas old dogs exhibited widespread staining for p-Tau
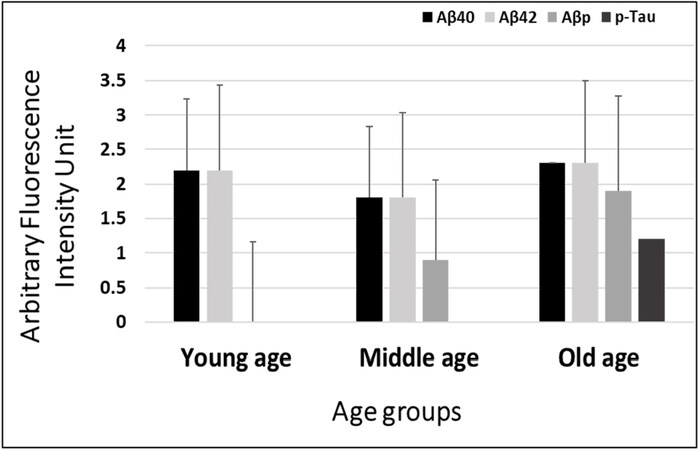
Figure 6. Semiquantitative analysis of Aβ40 and Aβ42 oligomers, Aβ plaques, and p-Tau in neurologically intact young (1-5-years), middle (6-10-years), and old (11-16-years) age groups of dogs. PRIOAD12 (Aβ40 oligomers), PRIOAD13 (Aβ42 oligomers), 4G8 (Aβp), and AT8 (p-Tau) fluorescence immunoreactivity and intensities were examined and quantified at 40× magnification under the fluorescence microscope. Semiquantitative analyses were compared with the three different age groups. A large amount of Aβ40 and Aβ42 oligomers were found in young dogs, which decreased in middle age group, then finally, an upward trend was noticed in older dogs. In comparison, Aβp were completely absent in young dogs, and then moderately found in middle, and large amounts in old age groups. Finally, p-Tau deposits were not found in young and middle age groups of dogs, whereas old dogs exhibited a considerable amount of p-Tau.
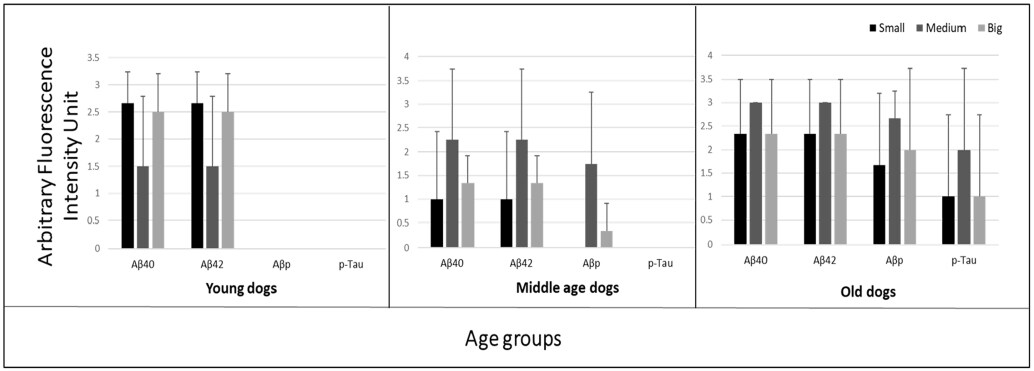
Figure 7. Influence of the size of dogs on the retinal deposition of amyloid-beta oligomers (Aβo), plaques (Aβp), and phosphorylated tau (p-Tau) in cognitively unimpaired young (1-5-years), middle (6-10-years) and old (11-16-years) age groups of dogs. PRIOAD12 (Aβ40 oligomers), PRIOAD13 (Aβ42 oligomers), 4G8 (Aβp), and AT8 (p-Tau) fluorescence immunoreactivity and intensities were examined and quantified at 40× magnification under the fluorescence microscope. In three different age groups, semiquantitative analyses were compared with the size of the dogs. In young dogs, a large number of oligomers were displayed by the small and big size dogs. The majority of medium-sized dogs displayed strong Aβo and Aβp staining intensity in the middle-aged dogs. Finally, among the different sizes of old dogs, medium-sized breeds displayed the highest amount of Aβo, Aβp, and p-Tau staining intensity.
Influence of eye pathology on the retinal deposition of Aβ oligomers, Aβ plaques, and phosphorylated tau in 30 dogs
Further, to understand whether the presence of pre-existing underlying eye pathology in the dogs may influence retinal Aβ and p-Tau depositions, we compared their staining intensity with the reported underlying clinical eye conditions [Table 2]. We found that four young dogs aged 1-5 years affected with anterior segment dysgenesis, a spectrum of disorders that affect the development of the anterior segment, including the cornea, iris, ciliary body, and lens[63-65], displayed "moderate" to "strong" intensity staining for Aβ40 and Aβ42 oligomers [Table 2]. However, another two dogs in this age group and affected with anterior segment dysgenesis showed little to no deposition of Aβ40 and Aβ42 oligomers [Table 2]. Eighteen out of thirty dogs presented with eye neoplasms, including two in the young age group, seven in the middle age group, and nine in the old age group [Table 2]. There was no clear relation between neoplasms and the intensity of Aβ or p-Tau [Table 2]. Interestingly, eye inflammation (proptosis/ phthisis bulbi in young dogs and conjunctivitis in a middle-aged dog) corresponded to Aβ40 and Aβ42 oligomers intensity that ranged from "moderate" to "strong" [Table 2]. Finally, a case of pre-glaucoma in the middle age group and a case of glaucoma in the old age group also matched well with Aβ40 and Aβ42 oligomers as well as Aβp intensity. Overall, this analysis appears to indicate that no direct influence of pre-existing eye disorders on Aβ and
DISCUSSION
Eye imaging can provide an opportunity to develop an easily accessible and point-of-care routine diagnostic testing to help predict MCI/AD early[66,67]. A study by Ko and colleagues examined the retinal NFL thickness and cognitive status of individuals aged 40 to 69 years over three years[68]. The authors found that individuals with thinner NFL showed a higher incidence of reduced cognition.
Despite the validation of the dog as a robust translational model for AD[24,30,69], only very limited studies investigated AD-related changes in the young and neurologically intact dogs. One study by Stylianaki and colleagues confirmed a similar pattern to that of humans; 61 dogs were subdivided into young (0-4 years old), middle-aged (4-8 years old), aged cognitively normal (8-20 years old), and aged cognitively impaired (8-17 years-old)[70]. The authors found that young dogs displayed the highest levels of total plasma Aβ42 and Aβ42/Αβ40 ratio and the middle-aged dogs had the highest cerebrospinal fluid Aβ40 and Aβ42 when compared to neurologically intact aged dogs. Our first step was to investigate the distribution and morphological appearance of both Aβ40 and Aβ42 oligomers representing the canine lifetime, in the retinal layers of these neurologically intact 30 dogs. Previous studies have shown that A11 anti-oligomer antibody binds to different epitopes of the Aβo but also reacts with oligomeric aggregates of other proteins independent of their primary sequences. Of note, A11 cannot differentiate between Aβ40 and Aβ42. In contrast, PRIOAD12 and PRIOAD13 nanobodies bind to Aβ40 and Aβ42, respectively. Previous studies reported an inverse interrelationship between neurotoxicity and the size of Aβo, where the toxic effect of Aβo was shown to decrease with increased size. The general molecular weight of Aβo was found to range between 10-100 kDa in AD brain and included dimers to dodecamer[71]. A study by Lambert and colleagues first describes the cytotoxic effect of the small diffusible Aβo on the hippocampal neurons[72]. The small Aβo referred to as Aβ-derived diffusible ligands toxicity was tested in organotypic mouse brain slice cultures. The authors found that 17 and 22 kDa small oligomers killed hippocampal neurons at nanomolar concentration[72]. In addition, a study by Cizas and colleagues confirmed that oligomers larger than 30 KDa have a less toxic effect on the inhibition of long-term potentiation[73]. They also suggested the transition of sizes from small to large correlate with the high to a low toxic effect of Aβo[73]. A11 specifically binds to the prefibrillar oligomers. However, studies suggested that the specificity of A11 to AD-related oligomers might vary as it recognized prefibrillar oligomers from various proteins that share a common structure including α-synuclein, islet amyloid polypeptide, polyglutamine (PolyQ), lysozyme, and prion peptide[36]. A study by Glabe and colleagues reported the ideal band size of prefibrillar oligomer-specific antibody A11 ranged from approximately tetrameric up to ~75 kDa[35]. In our study, we confirmed the presence of an A11-specific band at ~70KDa, confirming binding to prefibrillar oligomers in APP/PS1 mice. In comparison, camelid-derived single-domain nanobodies bind to ~15KDa band representing the small oligomers. In agreement with this current study, our previous report[55] also suggested that nanobodies were able to detect the toxic small diffusible oligomers specific to AD, whereas A11 binds to the larger oligomers believed to be less toxic to the neurons.
Studies suggested that in the canine brain, Aβo may be the toxic species responsible for cognitive decline and can potentially be an early biomarker for the detection of CCD[23,74]. Naaman and colleagues recently reported that Aβ42 oligomers cause extensive retinal neurotoxicity in rats when compared to fibrillary Aβ40 and Aβ42[75]. Our findings demonstrated extensive deposition of Aβ40 and Aβ42 oligomers in the retinal layers, including GCL, INL, and ONL in 26/30 in the young, middle, and old age groups, except for a 1.4-year-old male Siberian husky, a 7-year-old neutered male Hound mixed, an 8-year-old spayed female Bedlington terrier, and an 11-year-old neutered male mixed breed. Moreover, we did not notice any difference in the intensity of immunofluorescence (graded - to +++), the pattern of deposition, or morphology between Aβ40 and Aβ42 oligomers in all age groups. This presentation was unlikely influenced by the breed of the animals; for instance, out of three Siberian Huskies, one dog failed to display Aβ40 and Aβ42 oligomers accumulation. Remarkably, the younger age group, with an age range of 1-5 years, equivalent to humans aged 15-40 years according to the American Kennel Club[76], has displayed extensive deposition of Aβ oligomers. Of interest, a 2013 brain study investigated the presence of three different types of Aβ oligomers, including Aβ trimers, Aβ*56, and Aβ dimers, in 75 cognitively unimpaired individuals aged 1 to 96 years[14]. Young children and adolescents were positive for Aβ oligomers; the authors found an age-related accumulation of Aβ oligomers where the level of the amyloid-β dimer was significantly higher in subjects in their 60s, and amyloid-β trimer in their 70s, whereas Aβ*56 level was significantly higher in individuals in their 40s. The investigators proposed that Aβo, specifically Aβ*56, might trigger the pathological cascade in asymptomatic AD, a phase that may be identifiable two decades before the clinical onset[14].
Having found that the eyes of 26/30 dogs contained Aβ40 and Aβ42 oligomers, our next step was to demonstrate the presence/deposition of fibrillar Aβ in the canine retina of this same cohort. We show that none of the young dogs displays 4G8-positive Aβ fibrils and plaques (Aβp). However, Aβp were observed in some of the middle age group, where three dogs exhibited "low" staining intensity and two exhibited "strong" staining intensity in the GCL, IPL & INL. Aβp were also observed in the old age group, where five dogs showed "strong" signal intensity and two with "moderate" signal intensity in the GCL, IPL & INL. A pattern, not surprising, emerges; Aβ40 and Aβ42 oligomers are deposited in the retina following birth and subsequently, a fibrillar Aβ configuration, aggregated as plaques follow. Several studies have demonstrated the presence of Aβp in the canine brain and suggested only a weak correlation with the severity of cognitive dysfunction[77,78]. Schütt et al. investigated this question in further depth by studying dogs at differing ages, specifically aged 9-15 years, including neurologically intact dogs and dogs with CCD, and compared their amyloid burden with that of young dogs aged less than 6 years[69]. The authors reported that the levels of Aβ deposition strongly correlated with the age of the dog but not to their cognitive capacity. Asking the same questions regarding amyloid accretion in non-CNS neuronal populations, such as the retina, has not been investigated in the canine, but some recent reports confirmed their presence in the retina of AD[50,54,79]. In this study, we have demonstrated accumulation of retinal Aβp in the mature to an aged group of these 30 dogs which supports the hypothesis that Aβp might not influence the severity of cognitive deficits but can be a predictor of AD development[11,17]. Overall, our study revealed that the Aβ deposition pattern in the retina was such that Aβo was observed in all age groups, whereas Aβp accumulation was restricted to the middle and more intensely the old age group of dogs, regardless of demographic criteria, breed and gender. Interestingly, co-accumulation of both Aβo and Aβp were apparent in some middle and old dogs. This is in agreement with our report this year, where 17-18 months old APP/PS1 mice showed high levels of oligomers deposits in the retinal layers that co-localized with Aβp[55]. Previous neuropathological studies in AD have shown that cerebral Aβo is usually present in early disease stages and is the most cytotoxic of Aβ species, mostly responsible for neurotoxicity and synaptic dysfunction[34,80-83], whereas extracellular Aβp is believed to accumulate at later ages and may act as a reservoir for Aβo[84]. Moreover, the involvement of Aβo in retinal degeneration in AD has also been reported[85-87]. In the study reported herein, we noticed widespread distribution of Aβo in the retinal layers of dogs of all age groups, indicating potential involvement in retinal degeneration, perhaps leading to vision impairment in some dogs[88,89]. Of importance, a previous study by Ozawa and colleagues that focused on web and paper-based surveys of dogs aged ≥ 10 years to identify physical disturbances related to CCD showed that more than 90% of dogs affected with CCD had vision impairment[90]. In addition to the neural retina, we observed Aβ deposition s in the retinal microvasculature of young, middle, and old age groups, which might imitate CAA in AD[91-93]. Sharafi et al. suggested that retinal vasculature changes captured by hyperspectral imaging can differentiate cerebral amyloid status between cognitively impaired and unimpaired individuals[94].
Central to AD pathogenesis is the intraneuronal deposition of hyperphosphorylated tau (p-Tau) in granular form and eventual organization as NFTs, one of the cardinal neuropathological hallmarks[3,4]. In dogs with CCD, p-Tau, unlike NFTs, has been identified in the brain, but until recently, only in some cases. We speculate that, unlike human AD, p-Tau is only evident in pretangle granular form in dogs due to their shorter lifespan (Tayebi & Habiba, unpublished observation). p-Tau neuropathology was shown to develop about a decade before the formation of Aβp in AD brains and was hypothesized to trigger AD[95,96]. However, Aβo accumulation was shown to precede and drive p-Tau accumulation and transneuronal spread across synapses in the parietal cortex of AD[96].
In the retina of human AD patients, p-Tau displayed a diffuse pattern in the plexiform layers in the absence of NFTs[54]. Schön and colleagues reported the presence of AT8-positive intracellular NFTs and diffuse
While previous studies have demonstrated the presence of Aβp and p-Tau in the aged dog brain[30,43], Pugleise et al. proposed that the acquisition of diffuse Aβp and p-Tau are unrelated and independent events of aged dogs[42]. Whether these two “ingredients” of AD dementia or CCD are closely linked (co-localized or at least act in concert) is likely crucial. In AD, it was shown that p-Tau and Aβo cause neuronal toxicity and may act synergistically to trigger synaptic dysfunction[36,37,100]. Manczak and colleagues showed that Aβ40 or Aβ42 oligomers co-localized with p-Tau in the brains of AD patients[101]. The authors also reported that the interaction between Aβo and p-Tau became more prominent with disease progression and was more pronounced at Braak stage V and VI compared to Braak stage III and IV[102,103]. The evidence from this canine retina study would support an interaction: we also show that p-Tau co-localized with Aβ40 or Aβ42 oligomers in the OPL, INL, IPL, and GCL of the retina of 4/10 dogs in the eldest age group. A recent PET brain imaging study by Lockhart et al., which demonstrated the presence of both Aβ and Tau pathology in cognitively normal older adults, showed a significant spatial correlation with Aβ and Tau deposition[104]. Thus co-localization, while evident, does not itself necessarily imply neuronal cytotoxicity.
Several studies in the field of AD research highlighted the importance of diagnostic and therapeutic interventions in the asymptomatic phase while the individuals are cognitively intact[14,105]. However, it is challenging to first identify and then track disease progression in humans without sensitive, specific, and cost-effective early diagnostic approaches and a lack of robust natural translational models. In that context, dogs have the potential to be an effective natural model for the study of aging and AD. They share the same environment as humans[106,107]; there exists the option of brain CT and MRI imaging and intracranial biopsy, ease of cognitive testing which helps reduce physiological stress, and finally, a short lifespan[108]. Canine ophthalmology is a well-established clinical and investigative discipline (inherited retinopathies, for example). The current study provides strong impetus to track how this seemingly common process evolves, with potential consequences for aging, neurodegenerative disease, and also vision.
Lateralization in retinal and cerebral Aβ levels was previously investigated in an AD mouse model[109]. Some studies revealed left posterior brain-dominant lateralization in Aβ42/40; however, no left or right brain hemisphere or retinal dominance lateralization was observed for Aβ40 and Aβ42. Although our current study does not provide information about left versus right retinal accumulation of Aβ, the significance of the lateralization in human Alzheimer’s and CCD remains unknown. Moreover, previous reports have shown that Aβ accumulation was more prominent in the far-peripheral and mid-peripheral of the superior temporal quadrant than in the central retina in AD patients[50,79]; however, in our study, no notable differences in Aβ and tau deposition have been observed in different parts of the retina.
The canine eye tissues used in this study had been surgically removed in the course of managing spontaneous disorders of the globe, eyelids, or structures within the orbit which necessitated enucleation. It could be asked whether any of these conditions played any part in Aβ and/or p-Tau deposition in the retina. We think this unlikely, given the diverse clinical conditions involved, which ranged from developmental abnormalities, inflammatory conditions, and proptosis to various neoplasms of differing grades of the eye, while other cases were disorders arising in the conjunctiva or orbit. The span of ages in these dogs (from juvenile to quite aged) would further make this seem unlikely and some evidence of human retinal Aβ and/or p-Tau exists, though such explorations are largely performed only at the end of life.
DECLARATIONS
Authors’ contributionsPerformed experiments and revised manuscript: Habiba U
Revised manuscript: Morley J, Krockenberger M
Was a major contributor in writing and revising the manuscript: Summers BA
Conceived and designed experiments and wrote manuscript: Tayebi M
All authors read and approved the final manuscript.
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateSections of eyes used in this study were prepared from archived cases in the Comparative Ocular Pathology Laboratory of Wisconsin (COPLOW) at the Department of Pathobiological Sciences, School of Veterinary Medicine, University of Wisconsin, Madison. Such tissues, historically submitted for disease investigation purposes, are not subject to approval by institutional animal ethical regulations.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
Supplementary MaterialsREFERENCES
1. Clark CM, Karlawish JHT. Alzheimer disease: current concepts and emerging diagnostic and therapeutic strategies. Ann Intern Med 2003;138:400-10.
4. Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med 2011;1:a006189.
5. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 2002;297:353-6.
6. Jack CR Jr, Wiste HJ, Weigand SD, et al. Age, sex, and APOE ε4 effects on memory, brain structure, and β-amyloid across the adult life span. JAMA Neurol 2015;72:511-9.
7. Rentz DM, Locascio JJ, Becker JA, et al. Cognition, reserve, and amyloid deposition in normal aging. Ann Neurol 2010;67:353-64.
8. Rodrigue KM, Kennedy KM, Park DC. Beta-amyloid deposition and the aging brain. Neuropsychol Rev 2009;19:436-50.
9. Oh H, Mormino EC, Madison C, Hayenga A, Smiljic A, Jagust WJ. β-amyloid affects frontal and posterior brain networks in normal aging. Neuroimage 2011;54:1887-95.
10. Bourgeat P, Chételat G, Villemagne VL, et al. AIBL Research Group. Beta-amyloid burden in the temporal neocortex is related to hippocampal atrophy in elderly subjects without dementia. Neurology 2010;74:121-7.
11. Rowe CC, Ellis KA, Rimajova M, et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging 2010;31:1275-83.
12. Petersen RC, Aisen P, Boeve BF, et al. Mild cognitive impairment due to Alzheimer disease in the community. Ann Neurol 2013;74:199-208.
13. Erten-Lyons D, Woltjer RL, Dodge H, et al. Factors associated with resistance to dementia despite high Alzheimer disease pathology. Neurology 2009;72:354-60.
14. Lesné SE, Sherman MA, Grant M, et al. Brain amyloid-β oligomers in ageing and Alzheimer’s disease. Brain 2013;136:1383-98.
15. Bischof GN, Rodrigue KM, Kennedy KM, Devous MD Sr, Park DC. Amyloid deposition in younger adults is linked to episodic memory performance. Neurology 2016;87:2562-6.
16. Hanseeuw BJ, Betensky RA, Jacobs HIL, et al. Association of amyloid and tau with cognition in preclinical alzheimer disease: a longitudinal study. JAMA Neurol 2019;76:915-24.
17. Rowe CC, Bourgeat P, Ellis KA, et al. Predicting Alzheimer disease with β-amyloid imaging: results from the Australian imaging, biomarkers, and lifestyle study of ageing. Ann Neurol 2013;74:905-13.
18. Drummond E, Wisniewski T. Alzheimer’s disease: experimental models and reality. Acta Neuropathol 2017;133:155-75.
19. Kitazawa M, Medeiros R, Laferla FM. Transgenic mouse models of Alzheimer disease: developing a better model as a tool for therapeutic interventions. Curr Pharm Des 2012;18:1131-47.
20. Cummings BJ, Head E, Ruehl W, Milgram NW, Cotman CW. The canine as an animal model of human aging and dementia. Neurobiology of Aging 1996;17:259-68.
21. Head E, McCleary R, Hahn FF, Milgram NW, Cotman CW. Region-specific age at onset of β-amyloid in dogs. Neurobiol Aging 2000;21:89-96.
22. Pugliese M, Gangitano C, Ceccariglia S, et al. Canine cognitive dysfunction and the cerebellum: acetylcholinesterase reduction, neuronal and glial changes. Brain Res 2007;1139:85-94.
23. Head E, Pop V, Sarsoza F, et al. Amyloid-beta peptide and oligomers in the brain and cerebrospinal fluid of aged canines. J Alzheimers Dis 2010;20:637-46.
24. Youssef SA, Capucchio MT, Rofina JE, et al. Pathology of the aging brain in domestic and laboratory animals, and animal models of human neurodegenerative diseases. Vet Pathol 2016;53:327-48.
25. Cummings BJ, Head E, Afagh AJ, Milgram NW, Cotman CW. Beta-amyloid accumulation correlates with cognitive dysfunction in the aged canine. Neurobiol Learn Mem 1996;66:11-23.
26. Adams B, Chan A, Callahan H, Milgram NW. The canine as a model of human cognitive aging: recent developments. Prog Neuropsychopharmacol Biol Psychiatry 2000;24:675-92.
27. Head E. A canine model of human aging and Alzheimer’s disease. Biochim Biophys Acta 2013;1832:1384-9.
28. Rofina JE, van Ederen AM, Toussaint MJ, et al. Cognitive disturbances in old dogs suffering from the canine counterpart of Alzheimer’s disease. Brain Res 2006;1069:216-26.
29. Chow VW, Mattson MP, Wong PC, Gleichmann M. An overview of APP processing enzymes and products. Neuromolecular Med 2010;12:1-12.
30. Yu CH, Song GS, Yhee JY, et al. Histopathological and immunohistochemical comparison of the brain of human patients with Alzheimer’s disease and the brain of aged dogs with cognitive dysfunction. J Comp Pathol 2011;145:45-58.
31. Broersen K, Rousseau F, Schymkowitz J. The culprit behind amyloid beta peptide related neurotoxicity in Alzheimer’s disease: oligomer size or conformation? Alzheimers Res Ther 2010;2:12.
32. Goure WF, Krafft GA, Jerecic J, Hefti F. Targeting the proper amyloid-beta neuronal toxins: a path forward for Alzheimer’s disease immunotherapeutics. Alzheimers Res Ther 2014;6:42.
33. Morgado I, Fändrich M. Assembly of Alzheimer’s Aβ peptide into nanostructured amyloid fibrils. Current opinion in colloid & interface science 2011;16:508-14.
34. Sengupta U, Nilson AN, Kayed R. The role of amyloid-β oligomers in toxicity, propagation, and immunotherapy. EBioMedicine 2016;6:42-9.
36. Kayed R, Head E, Thompson JL, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003;300:486-9.
37. Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol 2007;8:101-12.
38. Benilova I, Karran E, De Strooper B. The toxic Aβ oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci 2012;15:349-57.
39. Zhao LN, Long HW, Mu Y, Chew LY. The toxicity of amyloid β oligomers. International journal of molecular sciences 2012;13:7303-27.
40. Abey A, Davies D, Goldsbury C, Buckland M, Valenzuela M, Duncan T. Distribution of tau hyperphosphorylation in canine dementia resembles early Alzheimer’s disease and other tauopathies. Brain Pathol 2021;31:144-62.
41. Papaioannou N, Tooten PC, van Ederen AM, et al. Immunohistochemical investigation of the brain of aged dogs. I. Detection of neurofibrillary tangles and of 4-hydroxynonenal protein, an oxidative damage product, in senile plaques. Amyloid 2001;8:11-21.
42. Pugliese M, Mascort J, Mahy N, Ferrer I. Diffuse beta-amyloid plaques and hyperphosphorylated tau are unrelated processes in aged dogs with behavioral deficits. Acta Neuropathol 2006;112:175-83.
43. Schmidt F, Boltze J, Jäger C, et al. Detection and quantification of β-amyloid, pyroglutamyl Aβ, and tau in aged canines. J Neuropathol Exp Neurol 2015;74:912-23.
44. Wegiel J, Wisniewski HM, Sołtysiak Z. Region- and cell-type-specific pattern of tau phosphorylation in dog brain. Brain Research 1998;802:259-66.
45. Armstrong RA. Alzheimer’s disease and the eye. Journal of Optometry 2009;2:103-11. Available from:
46. Santos CY, Johnson LN, Sinoff SE, Festa EK, Heindel WC, Snyder PJ. Change in retinal structural anatomy during the preclinical stage of Alzheimer’s disease. Alzheimers Dement (Amst) 2018;10:196-209.
47. Marquié M, Castilla-Martí M, Valero S, et al. Visual impairment in aging and cognitive decline: experience in a Memory Clinic. Sci Rep 2019;9:8698.
48. Lee ATC, Richards M, Chan WC, Chiu HFK, Lee RSY, Lam LCW. Higher dementia incidence in older adults with poor visual acuity. J Gerontol A Biol Sci Med Sci 2020;75:2162-8.
49. Habiba U, Merlin S, Lim JKH, et al. Age-specific retinal and cerebral immunodetection of amyloid-β plaques and oligomers in a rodent model of Alzheimer’s disease. J Alzheimers Dis 2020;76:1135-50.
50. Lee S, Jiang K, McIlmoyle B, et al. Amyloid Beta immunoreactivity in the retinal ganglion cell layer of the Alzheimer’s eye. Front Neurosci 2020;14:758.
51. Koronyo-Hamaoui M, Koronyo Y, Ljubimov AV, et al. Identification of amyloid plaques in retinas from Alzheimer’s patients and noninvasive in vivo optical imaging of retinal plaques in a mouse model. Neuroimage 2011;54 Suppl 1:S204-17.
52. Tsai Y, Lu B, Ljubimov AV, et al. Ocular changes in TgF344-AD rat model of Alzheimer’s disease. Invest Ophthalmol Vis Sci 2014;55:523-34.
53. Jentsch S, Schweitzer D, Schmidtke KU, et al. Retinal fluorescence lifetime imaging ophthalmoscopy measures depend on the severity of Alzheimer’s disease. Acta Ophthalmol 2015;93:e241-7.
54. den Haan J, Morrema THJ, Verbraak FD, et al. Amyloid-beta and phosphorylated tau in post-mortem Alzheimer’s disease retinas. Acta Neuropathol Commun 2018;6:147.
55. Habiba U, Descallar J, Kreilaus F, et al. Detection of retinal and blood Aβ oligomers with nanobodies. Alzheimers Dement (Amst) 2021;13:e12193.
56. Yi J, Chen B, Yao X, Lei Y, Ou F, Huang F. Upregulation of the lncRNA MEG3 improves cognitive impairment, alleviates neuronal damage, and inhibits activation of astrocytes in hippocampus tissues in Alzheimer’s disease through inactivating the PI3K/Akt signaling pathway. J Cell Biochem 2019;120:18053-65.
57. David MA, Jones DR, Tayebi M. Potential candidate camelid antibodies for the treatment of protein-misfolding diseases. J Neuroimmunol 2014;272:76-85.
58. van Eersel J, Stevens CH, Przybyla M, et al. Early-onset axonal pathology in a novel P301S-Tau transgenic mouse model of frontotemporal lobar degeneration. Neuropathol Appl Neurobiol 2015;41:906-25.
59. Radde R, Bolmont T, Kaeser SA, et al. Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep 2006;7:940-6.
60. Nakayama H, Uchida K, Doi K. A comparative study of age-related brain pathology - are neurodegenerative diseases present in nonhuman animals ? Med Hypotheses 2004;63:198-202.
61. Cummings BJ, Pike CJ, Shankle R, Cotman CW. β-amyloid deposition and other measures of neuropathology predict cognitive status in Alzheimer’s disease. Neurobiology of Aging 1996;17:921-33.
62. Breydo L, Uversky VN. Structural, morphological, and functional diversity of amyloid oligomers. FEBS Lett 2015;589:2640-8.
63. Silva EG, Dubielzig R, Zarfoss MK, Anibal A. Distinctive histopathologic features of canine optic nerve hypoplasia and aplasia: a retrospective review of 13 cases. Vet Ophthalmol 2008;11:23-9.
64. Rüttimann G, Daicker B. Complex colobomas in the anterior eye segment in a beagle hound. Zentralbl Veterinarmed A 1982;29:528-37.
66. Casaletto KB, Ward ME, Baker NS, et al. Retinal thinning is uniquely associated with medial temporal lobe atrophy in neurologically normal older adults. Neurobiol Aging 2017;51:141-7.
67. Alonso-Caneiro D, Read SA, Collins MJ. Automatic segmentation of choroidal thickness in optical coherence tomography. Biomed Opt Express 2013;4:2795-812.
68. Ko F, Muthy ZA, Gallacher J, et al. UK Biobank Eye & Vision Consortium. Association of retinal nerve fiber layer thinning with current and future cognitive decline: a study using optical coherence tomography. JAMA Neurol 2018;75:1198-205.
69. Schütt T, Helboe L, Pedersen LØ, Waldemar G, Berendt M, Pedersen JT. Dogs with cognitive dysfunction as a spontaneous model for early Alzheimer’s disease: a translational study of neuropathological and inflammatory markers. J Alzheimers Dis 2016;52:433-49.
70. Stylianaki I, Polizopoulou ZS, Theodoridis A, Koutouzidou G, Baka R, Papaioannou NG. Amyloid-beta plasma and cerebrospinal fluid biomarkers in aged dogs with cognitive dysfunction syndrome. J Vet Intern Med 2020;34:1532-40.
71. Kuo YM, Emmerling MR, Vigo-Pelfrey C, et al. Water-soluble abeta (N-40, N-42) oligomers in normal and Alzheimer disease brains. J Biol Chem 1996;271:4077-81.
72. Lambert MP, Barlow AK, Chromy BA, et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A 1998;95:6448-53.
73. Cizas P, Budvytyte R, Morkuniene R, et al. Size-dependent neurotoxicity of beta-amyloid oligomers. Arch Biochem Biophys 2010;496:84-92.
74. Rusbridge C, Salguero FJ, David MA, et al. An aged canid with behavioral deficits exhibits blood and cerebrospinal fluid amyloid beta oligomers. Front Aging Neurosci 2018;10:7.
75. Naaman E, Ya’ari S, Itzkovich C, et al. The retinal toxicity profile towards assemblies of Amyloid-β indicate the predominant pathophysiological activity of oligomeric species. Sci Rep 2020;10:20954.
76. American Kennel Club. Dog years to human years 2018, January 02. Available from: https://www.akcpetinsurance.com/blog/year-of-the-dog [Last accessed on 23 May 2022].
77. Ozawa M, Chambers JK, Uchida K, Nakayama H. The relation between canine cognitive dysfunction and age-related brain lesions. J Vet Med Sci 2016;78:997-1006.
78. Kiatipattanasakul W, Nakamura S, Hossain MM, et al. Apoptosis in the aged dog brain. Acta Neuropathol 1996;92:242-8.
79. Koronyo Y, Biggs D, Barron E, et al. Retinal amyloid pathology and proof-of-concept imaging trial in Alzheimer’s disease. JCI Insight 2017;2:93621.
80. El-Agnaf OM, Salem SA, Paleologou KE, et al. Detection of oligomeric forms of alpha-synuclein protein in human plasma as a potential biomarker for Parkinson’s disease. FASEB J 2006;20:419-25.
81. El-agnaf OM, Walsh DM, Allsop D. Soluble oligomers for the diagnosis of neurodegenerative diseases. The Lancet Neurology 2003;2:461-2.
82. Mucke L, Selkoe DJ. Neurotoxicity of amyloid β-protein: synaptic and network dysfunction. Cold Spring Harb Perspect Med 2012;2:a006338.
83. Kim HJ, Chae SC, Lee DK, et al. Selective neuronal degeneration induced by soluble oligomeric amyloid beta protein. FASEB J 2003;17:118-20.
85. Lynn SA, Johnston DA, Scott JA, et al. Oligomeric Aβ1-42 induces an AMD-like phenotype and accumulates in lysosomes to impair RPE function. Cells 2021;10:413.
86. Ratnayaka JA, Serpell LC, Lotery AJ. Dementia of the eye: the role of amyloid beta in retinal degeneration. Eye (Lond) 2015;29:1013-26.
87. Luibl V, Isas JM, Kayed R, Glabe CG, Langen R, Chen J. Drusen deposits associated with aging and age-related macular degeneration contain nonfibrillar amyloid oligomers. J Clin Invest 2006;116:378-85.
88. Denenberg S, Liebel F, Rose J. Behavioural and medical differentials of cognitive decline and dementia in dogs and cats. In: Landsberg G, Maďari A, Žilka N, editors. Canine and feline dementia. Cham: Springer International Publishing; 2017. p. 13-58.
89. Acland G, Aguirre G. Retinal degenerations in the dog: IV. Early retinal degeneration (erd) in Norwegian elkhounds. Exp Eye Res 1987;44:491-521.
90. Ozawa M, Inoue M, Uchida K, Chambers JK, Takeuch Y, Nakayama H. Physical signs of canine cognitive dysfunction. J Vet Med Sci 2019;81:1829-34.
91. Weber SA, Patel RK, Lutsep HL. Cerebral amyloid angiopathy: diagnosis and potential therapies. Expert Rev Neurother 2018;18:503-13.
92. Patton N, Aslam T, Macgillivray T, Pattie A, Deary IJ, Dhillon B. Retinal vascular image analysis as a potential screening tool for cerebrovascular disease: a rationale based on homology between cerebral and retinal microvasculatures. J Anat 2005;206:319-48.
93. Shi H, Koronyo Y, Rentsendorj A, et al. Identification of early pericyte loss and vascular amyloidosis in Alzheimer’s disease retina. Acta Neuropathol 2020;139:813-36.
94. Sharafi SM, Sylvestre JP, Chevrefils C, et al. Vascular retinal biomarkers improves the detection of the likely cerebral amyloid status from hyperspectral retinal images. Alzheimers Dement (N Y) 2019;5:610-7.
95. Arnsten AFT, Datta D, Del Tredici K, Braak H. Hypothesis: tau pathology is an initiating factor in sporadic Alzheimer’s disease. Alzheimers Dement 2021;17:115-24.
96. Bilousova T, Miller CA, Poon WW, et al. Synaptic amyloid-β oligomers precede p-Tau and differentiate high pathology control cases. Am J Pathol 2016;186:185-98.
97. Schön C, Hoffmann NA, Ochs SM, et al. Long-term in vivo imaging of fibrillar tau in the retina of P301S transgenic mice. PLoS One 2012;7:e53547.
98. Rofina J, van Andel I, van Ederen AM, Papaioannou N, Yamaguchi H, Gruys E. Canine counterpart of senile dementia of the Alzheimer type: amyloid plaques near capillaries but lack of spatial relationship with activated microglia and macrophages. Amyloid 2003;10:86-96.
99. Colle MA, Hauw JJ, Crespeau F, et al. Vascular and parenchymal abeta deposition in the aging dog: correlation with behavior. Neurobiol Aging 2000;21:695-704.
100. De Felice FG, Wu D, Lambert MP, et al. Alzheimer’s disease-type neuronal tau hyperphosphorylation induced by A beta oligomers. Neurobiol Aging 2008;29:1334-47.
101. Manczak M, Reddy PH. Abnormal interaction of oligomeric amyloid-β with phosphorylated tau: implications to synaptic dysfunction and neuronal damage. J Alzheimers Dis 2013;36:285-95.
102. Fein JA, Sokolow S, Miller CA, et al. Co-localization of amyloid beta and tau pathology in Alzheimer’s disease synaptosomes. Am J Pathol 2008;172:1683-92.
103. Takahashi RH, Capetillo-Zarate E, Lin MT, Milner TA, Gouras GK. Co-occurrence of Alzheimer’s disease β-amyloid and τ pathologies at synapses. Neurobiol Aging 2010;31:1145-52.
104. Lockhart SN, Schöll M, Baker SL, et al. Amyloid and tau PET demonstrate region-specific associations in normal older people. Neuroimage 2017;150:191-9.
105. Golde TE, Schneider LS, Koo EH. Anti-aβ therapeutics in Alzheimer’s disease: the need for a paradigm shift. Neuron 2011;69:203-13.
106. Parker HG, Kim LV, Sutter NB, et al. Genetic structure of the purebred domestic dog. Science 2004;304:1160-4.
107. Davis PR, Head E. Prevention approaches in a preclinical canine model of Alzheimer’s disease: benefits and challenges. Front Pharmacol 2014;5:47.
108. Awano T, Johnson GS, Wade CM, et al. Genome-wide association analysis reveals a SOD1 mutation in canine degenerative myelopathy that resembles amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 2009;106:2794-9.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Habiba U, Morley J, Krockenberger M, Summers BA, Tayebi M. A sequential deposition of amyloid beta oligomers, plaques and phosphorylated tau occurs throughout life in the canine retina. Ageing Neur Dis 2022;2:7. http://dx.doi.org/10.20517/and.2022.06
AMA Style
Habiba U, Morley J, Krockenberger M, Summers BA, Tayebi M. A sequential deposition of amyloid beta oligomers, plaques and phosphorylated tau occurs throughout life in the canine retina. Ageing and Neurodegenerative Diseases. 2022; 2(2): 7. http://dx.doi.org/10.20517/and.2022.06
Chicago/Turabian Style
Habiba, Umma, John Morley, Mark Krockenberger, Brian A. Summers, Mourad Tayebi. 2022. "A sequential deposition of amyloid beta oligomers, plaques and phosphorylated tau occurs throughout life in the canine retina" Ageing and Neurodegenerative Diseases. 2, no.2: 7. http://dx.doi.org/10.20517/and.2022.06
ACS Style
Habiba, U.; Morley J.; Krockenberger M.; Summers BA.; Tayebi M. A sequential deposition of amyloid beta oligomers, plaques and phosphorylated tau occurs throughout life in the canine retina. Ageing. Neur. Dis. 2022, 2, 7. http://dx.doi.org/10.20517/and.2022.06
About This Article
Copyright

Data & Comments
Data


 Cite This Article 13 clicks
Cite This Article 13 clicks

 Like This Article 35
likes
Like This Article 35
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.