
Review of evidence implicating the plasminogen activator system in blood-brain barrier dysfunction associated with Alzheimer's disease

Abstract
Elucidating the pathogenic mechanisms of Alzheimer’s disease (AD) to identify therapeutic targets has been the focus of many decades of research. While deposition of extracellular amyloid-beta plaques and intraneuronal neurofibrillary tangles of hyperphosphorylated tau have historically been the two characteristic hallmarks of AD pathology, therapeutic strategies targeting these proteinopathies have not been successful in the clinics. Neuroinflammation has been gaining more attention as a therapeutic target because increasing evidence implicates neuroinflammation as a key factor in the early onset of AD disease progression. The peripheral immune response has emerged as an important contributor to the chronic neuroinflammation associated with AD pathophysiology. In this context, the plasminogen activator system (PAS), also referred to as the vasculature’s fibrinolytic system, is emerging as a potential factor in AD pathogenesis. Evolving evidence suggests that the PAS plays a role in linking chronic peripheral inflammatory conditions to neuroinflammation in the brain. While the PAS is better known for its peripheral functions, components of the PAS are expressed in the brain and have been demonstrated to alter neuroinflammation and blood-brain barrier (BBB) permeation. Here, we review plasmin-dependent and -independent mechanisms by which the PAS modulates the BBB in AD pathogenesis and discuss therapeutic implications of these observations.
Keywords
INTRODUCTION
Alzheimer’s disease (AD) is recognized as the most common cause of dementia in the elderly, and over 6 million Americans are currently living with this disorder. In the United States, AD is the sixth leading single cause of death and the second most common contributing cause of death. The hallmark neuropathologic characteristic of AD is abnormal extracellular protein accumulation in the brain, notably the extracellular deposition of amyloid-β (Aβ) peptide generated from the improper cleavage of amyloid precursor protein (APP) that gives rise to Aβ monomers that aggregate into oligomeric Aβ fibrils and plaques, and intraneuronal neurofibrillary tangles (NF) comprised largely of hyperphosphorylated tau. These proteinopathies are associated with the loss of synapses and subsequent neuronal cell loss in the entorhinal cortex, hippocampus, and frontal cortex[1-3], and currently, the biomarkers most commonly used in human AD studies are beta-amyloid 42, tau, and phospho-tau proteins in the cerebrospinal fluid. More recently, blood p-tau181 has been reported as being a useful biomarker for distinguishing AD from other dementias[4]. Thus, it has been widely posited that Aβ plaques and/or abnormal hyperphosphorylated tau protein accumulation are causally linked to the behavioral and neurologic symptoms of AD. However, therapeutic strategies for decreasing Aβ plaque load[5,6], reducing Aβ production with BACE-1 inhibitors[7], or inhibiting hyperphosphorylated tau aggregation[8], have been largely unsuccessful in clinical trials over the past several years[3]. These failed clinical trials coupled with observations of age-related increases in Aβ deposition in cognitively intact individuals as well as evidence that Aβ plaque load does not closely correspond with cognitive decline in AD patients[1,9] and neurofibrillary tangles are associated with severe cognitive impairment characteristic of late stages of AD[10,11], have prompted research into alternative pathogenic mechanisms of AD.
It is now recognized that the extracellular deposition of Aβ and hyperphosphorylated tau triggers pro-inflammatory responses in microglia and astrocytes[12-14]. The neuroinflammatory response in AD has been described in detail in several recent reviews[14,15], and it appears that neuroinflammation plays an important role in the early progression of AD[16,17]. Multiple investigators have shown that Aβ monofibrils, oligomers, and plaques activate gene expression of pro-inflammatory mediators in microglia and astrocytes[13,16,18,19]. While microglial phagocytosis of amyloid may be neuroprotective in the early stages of AD by promoting
While the initial focus on the role of the immune response in AD pathogenesis has been on the brain’s intrinsic neuroinflammatory response, attention is now being directed to multiple systemic inflammatory disorders that accelerate or in some instances may be the primary trigger for neuroinflammatory responses that initiate and/or promote AD and other dementias[31-34]. Some of the observations that have stimulated this shift in focus include reports that young children chronically exposed to high levels of air pollution were found to have neuropathological hallmarks of AD upon incidental autopsy[35,36], and evidence that type 2 diabetes/ metabolic syndrome and inflammatory bowel disease are associated with increased risk of developing AD[15,37,38]. The causal factors linking peripheral inflammatory conditions to AD are likely multi-factorial and have not yet been clearly delineated; however, several mechanisms are emerging. Peripheral inflammatory conditions have been shown to (1) generate inflammatory cytokines that facilitate access of peripheral inflammatory lymphocytes into the CNS, most notably TNFα, IL-1β, and IL-6; (2) cause dysfunction of the blood-brain barrier (BBB); and (3) activate the plasminogen activator system (PAS), which has direct effects on the CNS and further facilitates BBB dysfunction. The remainder of this review will investigate the role of the PAS in mediating inflammatory crosstalk between the periphery and the brain and its potential role in AD pathogenesis.
PLASMINOGEN ACTIVATOR SYSTEM
The plasminogen activator system (PAS) is comprised of a group of serine proteases, inhibitors, and binding proteins that control the activity of the serine protease plasmin [Figure 1][39]. Plasmin plays a key role in the fibrinolysis cascade, catalyzing the final degradation of fibrin and various extracellular matrix proteins[40,41]. The zymogen plasminogen (PlG) is converted to activated plasmin by plasmin activators, which include tissue-type plasminogen activator (tPA) and urokinase-type plasminogen activator (uPA). tPA is primarily involved in intravascular fibrinolysis, activating plasminogen that is bound to polymerized fibrin. In contrast, uPA is secreted as a pro-enzyme whose active form is primarily localized on cell surfaces where it binds to the uPA receptor (uPAR). Plasminogen conversion by tPA and uPA in both the periphery and the CNS is tightly regulated by serine protease inhibitors (serpins). Serpins represent a superfamily of proteins with similar structures. Most relevant to this discussion are plasminogen activator inhibitor type 1 (PAI-1) and neuroserpin (NSP). PAI-1 irreversibly inhibits uPA or tPA by undergoing a large conformational change upon binding uPA or tPA that disrupts the active site of the plasmin activator and of PAI-1. In contrast, NSP preferentially inhibits tPA by forming an unstable complex that can release active tPA[42]. Reflecting the need for stringent regulation of the plasminogen cascade, free forms of activated plasmin activators, PAI-1, and NSP exist at very low concentrations with half lives in the order of minutes[43,44].
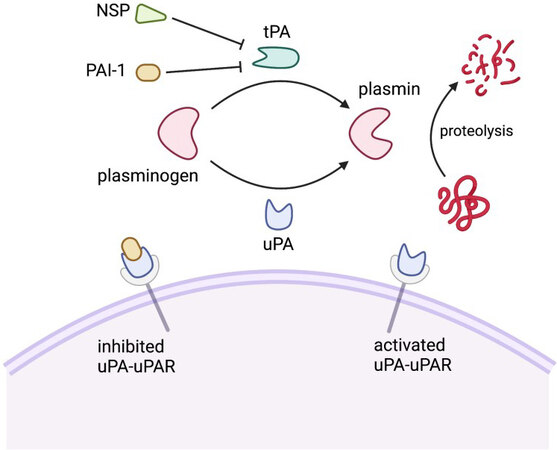
Figure 1. Schematic diagram of the molecular mechanisms of the plasminogen activator system. PAI-1: Plasminogen activator
PAS in the periphery
The peripheral PAS plays a central role in mediating fibrinolysis, extracellular migration, cell signaling, cellular migration, and tumor growth, which has been reviewed in detail elsewhere[45,46]. The PAS converts inactive plasminogen to plasmin, a trypsin-like serine protease, via the catalytic activity of PA[41]. Plasminogen is primarily present in platelets in the plasma and liver. However, in mice, plasminogen mRNA has been found in the adrenal, kidney, brain, testis, heart, lung, uterus, spleen, thymus, and gut[40,47]. In the periphery, PAI-1 serves as the main suppressor of plasma fibrinolytic activity[40]. In the bloodstream, PAI-1 exists on its own in an active form, or as part of a complex with tPA or vitronectin, a glycoprotein that can convert PAI-1 into its active form. Elevated levels of PAI-1 are associated with metabolic syndrome and associated with increased risk of atherothrombosis and stroke[48,49].
PAS in the CNS
In the CNS, plasminogen is expressed at low levels by neurons in the hippocampus, cortex, cerebellum, as well as neuroendocrine tissues, but it is primarily transported to the brain via systemic circulation[12,50,51]. Plasminogen has been localized to the extracellular space, while the plasmin activators, tPA and uPA, have been localized to not only the extracellular space, but also to neurons and astrocytes. Both plasmin activators have been shown to modulate synaptic function when released into the synaptic cleft[52-54]. Membrane depolarization induces the rapid release of tPA from cerebral cortical neurons, which modulates neuronal plasticity, learning, stress-induced anxiety, and visual cortex plasticity[55]. tPA and uPA activities have been localized to well-defined areas of the brain[56-59] and shown to participate in intracellular signaling that is independent of plasminogen activation (see below). tPA is the principal plasmin activator in the CNS with PAI-1 regulating its activity primarily in the extracellular space. NSP is primarily localized in neurons in the developing brain with very low levels detected in the mature CNS[60], where it preferentially binds to and inhibits tPA[61]. Interestingly, mutations of NSP are associated with rare familial dementia characterized by neuronal inclusion bodies that are biochemically comprised of polymers of NSP[62].
Plasmin activity has been shown to be upregulated in axonal growth and synaptic pruning, suggesting a role in brain development and regeneration that is not yet well understood[50]. While both tPA and uPA can mediate plasminogen activation in the CNS, plasminogen activation is primarily controlled by the tight regulation between tPA and PAI-1[51]. uPA has a low baseline expression in specific neurons and astrocytes in the normal brain, but is upregulated in pathologically inflammatory environments, such as multiple sclerosis and epilepsy[50,51]. Endothelial cells of microvessels in the brain contribute to the production of tPA, but tPA can also be expressed by glial cells, neurons, and infiltrating leukocytes, implicating a broad spectrum of tPA involvement in the brain. While tPA in the mature brain is detected primarily in neurons, its enzymatic activity is primarily restricted to the hippocampus, amygdala and hypothalamus[63,64]. The discrepancy between the expression of tPA mRNA and its areas of enzymatic activity is consistent with its trafficking and transport to mossy fiber tracts[63,64].
The plasmin activators, tPA and uPA have been shown to play an important role in CNS function and dysfunction with some of their functions being independent of plasminogen[65,66]. Extracellular tPA participates in cerebellar motor learning[67], remodeling in various nonneural tissues[67], and neuronal regeneration following ischemic injury[68]. tPA also participates in the regulation of BBB permeability[69,70]. Neuronal uPA is present in lower levels than tPA, participating in neurogenesis in the developing brain[71]. Its release in the mature central nervous system triggers astrocytic activation[53] and, like tPA, uPA promotes axonal and synaptic recovery following different forms of injury[72]. Both tPA and uPA are found in pre-synaptic vesicles that are released by calcium-dependent mechanisms[52,54,55].
The PAS is altered in AD
There has been longstanding interest in the role of the PAS in AD beginning with early reports that active plasmin efficiently digests Aβ peptides[73-77] both in vitro and in rodent AD models[19,73,74,76-81]. In the AD brain, tPA is highly expressed in regions of AD plaques, and in AD models where tPA is genetically inactivated, there is an increased accumulation of Aβ, synaptic dysfunction and memory deficits[78]. However, the enzymatic ability of brain tPA and uPA to activate plasmin in vivo is thought to be prevented by irreversible binding to high levels of extracellular PAI-1 secreted by immune-activated microglia and astrocytes[18]. PAI-1 is minimally expressed in the normal brain or cerebral vasculature, but does increase with senescence[82-84]. Brain levels of PAI-1 are also markedly increased in APP/PS1 mice[66] and the serum levels of PAI-1 are positively correlated with cognitive impairment in AD patients[85]. Consistent with the hypothesis that PAI-1 promotes AD pathology, genetic knockdown or small molecule inhibitors of PAI-1 reduced plaque formation in AD rodent models, and the small molecule PAI-1 inhibitor, PAZ-417, was shown to significantly improve hippocampal LTP and cognitive function in AD mice[73,74,86,87]. This finding was confirmed recently in an APP/PS1 AD mouse model using another small molecule PAI-1 inhibitor[86].
Whether tPA primarily plays a beneficial or detrimental role in AD progression is debated. Several studies have demonstrated that tPA activation of plasmin enzymatically reduces Aβ accumulation[78]. Conversely, tPA has been shown to mediate excitotoxic neurodegeneration by activating plasmin and causing subsequent laminin degradation[66,78]. Independent of plasmin activation, tPA causes GSK3 activation, tau hyperphosphorylation, microtubule destabilization, and neurotoxicity in rodent hippocampal neurons[88]. It has also been shown to mediate amyloid-induced microglial activation[89]. Based on such observations, it has been proposed that tPA contributes to neurotoxicity, microglial activation, and tau hyperphosphorylation as part of a feed-forward inflammatory pathway[73,88,89].
PAI-1 expression has been reported to be increased in the plasma[85,90,91] and brain tissues of AD patients[76]. PAI-1 expression is not detected in normal healthy human brains but is sporadically present in aged brains[84,92], and possibly linked to cerebrovascular disease. PAI-1 is the primary regulator of tPA in the CNS extracellular space and is a proinflammatory biomarker. Cytokines upregulate PAI-1 expression in microglia and astrocytes in human and animal models of AD[18,93]. The PAI-1 promoter is activated by
Congophilic amyloid angiopathy (CAA) is a vascular complication of AD in which Aβ40 plaques accumulate within the brain endothelium of cerebral arteries, arterioles and capillaries[101]. CAA can result in intracranial hemorrhages, cognitive impairment, or subacute inflammatory encephalopathy. tPA activation of endothelial NMDA receptors has been shown to regulate neurovascular coupling via nitric oxide-mediated regulation of cerebral blood flow. Elevated levels of brain PAI-1 impairs this tPA-dependent neurovascular coupling in Tg2576 AD mice, and pharmacologic inhibition of PAI-1 was shown to improve cognition in this animal model by selectively restoring neurovascular function while not affecting cortical amyloid plaques[102].
PAS modulates BBB integrity in AD
There is increasing evidence identifying BBB leakage as an early sign of cognitive dysfunction, as well as evidence linking BBB dysfunction to AD pathogenesis[103,104] and its neuroinflammatory pathology[33,105]. However, the mechanisms underlying BBB dysfunction in AD are currently not well-elucidated. The BBB is part of the neurovascular unit (NVU) in the brain, which consists of endothelial cells (ECs), mural cells, including vascular smooth muscle cells and pericytes, basement membrane, glia cells including astrocytes and microglia, and neurons [Figure 2]. The ECs of the BBB are a distinct characteristic of the NVU due to their tight junctions and lack of fenestrae. This allows the ECs to regulate the selective transport and metabolism of substances from blood to brain and vice versa, thereby separating the microenvironment of the brain parenchyma from changes in circulating ion and metabolite concentrations in the systemic circulation[105].
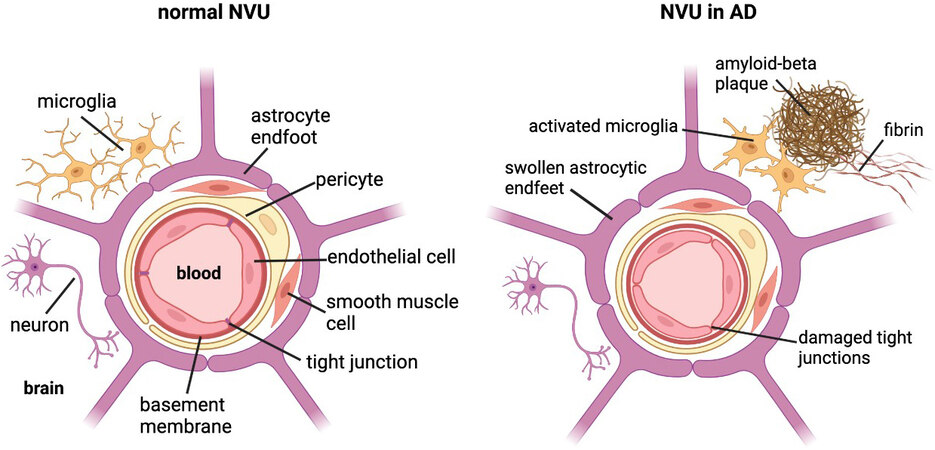
Figure 2. Cross-section of the neurovascular unit (NVU) in a normal brain vs. an Alzheimer’s disease (AD) brain. The blood-brain barrier (BBB) consists of endothelial cells joined by tight junctions, basement membrane, mural cells (i.e., pericytes and vascular smooth muscle cells), enclosed by astrocytic endfeet. Neurons and microglia closely associate with the BBB. In the AD brain, the NVU undergoes morphological and structural changes due to AD pathology. Amyloid-beta plaques complexed to fibrin result in neuroinflammation and BBB disruption, including activated microglia, swollen astrocytic endfeet, and compromised tight junctions. Created with BioRender.com.
In CNS injury, there are several potential mechanisms by which tPA is able to mediate changes in the permeability of the BBB [Figure 3], which in turn further exacerbates CNS injury by promoting neuroinflammation. AD is associated with BBB dysfunction in humans and animal models. Amyloid deposition activates gliosis that can alter the morphology of astrocytic endfeet, which are integral to the integrity of the neurovascular unit. As described previously with CAA, amyloid deposition can also injure the brain endothelium, which can additionally impair BBB integrity[106]. Finally, Aβ oligomers stimulate fibrin production that complexes with amyloid plaques, and fibrin has been shown to be increased in the parenchyma and vasculature of AD brains[107,108]. This fibrin-Aβ complex promotes further neuroinflammation and neurodegeneration. tPA is conformationally activated by fibrin deposition, but its enzymatic activity is inhibited by the elevated levels of PAI-1 found in AD parenchyma. However, as summarized in Figure 3, activated tPA has multiple plasmin-independent mechanisms by which it can compromise BBB integrity.
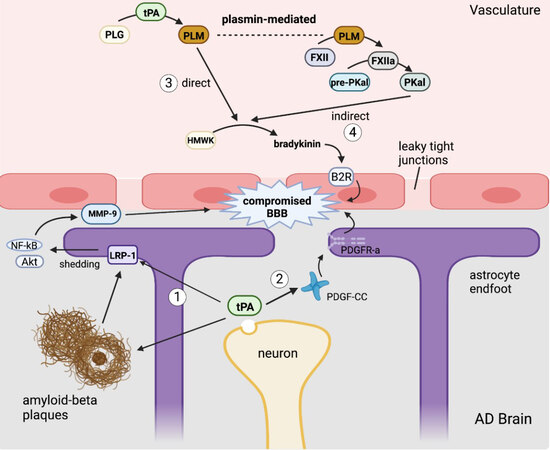
Figure 3. Mechanisms by which tPA may disrupt the blood-brain barrier. (1) tissue-type plasminogen activator(tPA) released from neurons cleaves lipoprotein receptor-related protein-1 (LRP-1) to activate an NF-κB signaling cascade resulting in the production of MMP-9. tPA and LRP-1 can bind amyloid beta, which facilitates Aβ endocytosis across the blood-brain barrier (BBB). (2) Neuronal tPA degrades platelet-derived growth factor-CC (PDGF-CC) to release the active ligand for PDGF receptor-α (PDGFR-α) on astrocytic endfeet, causing them to retract from endothelial cells. (3) Plasma tPA activates plasmin to directly produce bradykinin that activates bradykinin 2 receptor (B2R) receptor on endothelial cells. (4) Plasma tPA cleaves plasminogen to generate plasmin that indirectly upregulates bradykinin expression through plasma kallikrein (PKal). Created with BioRender.com.
tPA in the CNS directly alters BBB integrity
tPA has long been known to play a significant role in the NVU, mostly in the context of stroke[109-111]. tPA has been reported to directly alter the BBB integrity by triggering activation of LRP-1 on the surface of astrocytes[12]. LRP-1 is a multifunctional signaling receptor that functions in receptor-mediated endocytosis and cellular signaling. LRP-1 binds many ligands, including tPA and amyloid-beta[112], which thereby facilitates Aβ endocytosis across endothelial cells of the BBB[113]. Aβ oligomers may compromise BBB integrity via activation of matrix metalloproteinases (MMPs)[113]. Alternatively, tPA may cleave LRP-1 at its substrate binding ectodomain, activating NF-κB, which promotes the synthesis of matrix metalloproteinases MMP-3 and MMP-9, leading to matrix protein degradation and BBB leakage[12]. tPA-induced activation of LRP-1 shedding from astrocytic endfeet also promotes detachment of endfeet projections from tight junctions of the endothelial cells of the neurovascular unit, further compromising the BBB[12]. Additionally, tPA can directly alter BBB integrity via platelet-derived growth factor PDGF-CC[114]. Upregulated neuronal expression of tPA expression induced by CNS disease or injury results in the release of tPA into the extracellular matrix of the brain, where it cleaves complement subcomponents C1r/C1s, urchin EGF-like protein, and bone-morphogenic protein-1 (CUB) from PDGF-CC forming an active ligand that binds to PDGF receptor-α (PDGFR-α). PDGFR-α promotes BBB leakage that worsens cerebral edema, neuroinflammation and neuronal death[114]. One study found this tPA-mediated activation of PDGF-CC to be inefficient in an in vitro stroke model[115]. However, in vivo, the
Peripheral tPA alters BBB
In addition to its endogenous effects within the CNS, peripheral tPA can cross the intact BBB[116], phosphorylate claudin-5 and occludin, thereby weakening endothelial tight junctions and increasing BBB permeability by plasmin-independent mechanisms[117,118]. Chronic release of plasma tPA can induce a hyperfibrinolytic state that also directly increases vascular permeability of the BBB. Resultant plasmin activation by tPA also triggers bradykinin (BK) production[119,120]. BK is a peptide mediator generated from its circulating precursor, high molecular weight kininogen (HMWK), and is known for its ability to induce vascular permeability and cause vasodilation of arteries and veins[119]. It is a pro-inflammatory mediator, and its role as a neuromediator was identified in clinical conditions including AD[119]. While it is still debated as to how the PAS triggers BK generation, two primary pathways have been proposed [Figure 3]. A direct mechanism identified using an in vitro model involves tPA-mediated conversion of plasminogen to plasmin, which then cleaves HMWK into BK. BK acts through the bradykinin 2 receptor (B2R) on endothelial cells, triggering a signaling cascade that promotes intracellular calcium release and downregulation of claudin-5, a critical protein involved in maintaining EC tight junctions[120]. B2R activation can additionally induce tPA release from endothelial cells, further amplifying additional BK generation[121]. The PAS alternatively can indirectly trigger BK formation through a plasmin-dependent pathway where plasmin activated by tPA then converts Factor XII (FXII) into Factor XIIa (FXIIa), which then converts plasma pre-kallikrein into plasma kallikrein (PKal)[121]. PKal then serves to cleave HMWK, leading to BK formation and B2R signaling activation [Figure 3]. This indirect mechanism was demonstrated ex vivo and in vivo with the former using human plasma incubated with tPA, which resulted in the formation of active PKal; the latter demonstrating that intravenous injection of tPA in mice increased PKal activity[121,122].
AD has been shown to produce BBB dysfunction in humans and animal models. Amyloid deposition activates gliosis that can alter the morphology of astrocytic endfeet, which are integral to the integrity of the neurovascular unit. As described previously with CAA, amyloid deposition can injure the brain endothelium, which can additionally impair BBB integrity[106]. Finally, Aβ oligomers stimulate fibrin production that complexes with amyloid plaques and has been shown to be increased in the parenchyma and vasculature of AD brains[107]. This fibrin-Aβ complex promotes further neuroinflammation and neurodegeneration. tPA is conformationally activated by fibrin deposition, but its enzymatic activity is inhibited by the elevated levels of PAI-1 found in AD parenchyma. However, as summarized in Figure 3, activated tPA has multiple plasmin-independent mechanisms by which it can compromise BBB integrity.
CONCLUSION
Over the past two decades following initial reports of histologic evidence of Aβ deposition in the brains of children chronically exposed to severe air pollution[123], it has become clear that chronic peripheral inflammatory conditions, including those that involve lung, gut, liver, and metabolic syndrome, exacerbate or initiate neuroinflammatory disorders. This has been supported by epidemiologic findings of a positive association between chronic peripheral inflammatory conditions and increased incidence of dementia, including AD. More recently, there has been increased interest in the contribution of the peripheral PAS to the neuroinflammatory component of AD. Recently, it has become recognized that the risk of blood clots, increased mortality, and persistent neuroinflammatory complications of COVID 19 viral infections are also associated with pre-existing systemic inflammatory disorders shown to chronically activate components of the PAS[124]. With respect to AD, the available evidence suggests that the peripheral PAS may modulate the neuroinflammatory response via multiple mechanisms[12,51]. Besides fostering the transcytosis of inflammatory cells across the BBB, components of the PAS have been shown to decrease BBB integrity and increase BBB permeability, consequences that have been independently linked to early cognitive dysfunction[125] including progressive stages of AD[126] perhaps in association with concomitant vascular disease[127]. Overall, the means by which the PAS modulates BBB integrity by tPA and plasmin-dependent mechanisms is complex and requires further validation and investigation. tPA in the CNS has been shown to alter BBB permeability by LRP-1 and PDGF-CC-dependent mechanisms, while tPA produced from peripheral inflammation can cross the BBB where it may work in tandem with the kinin system to directly generate BK via plasmin, or indirectly by increased PKal. It is likely that tPA works multifactorially and that these mechanisms are not mutually exclusive [Figure 2][118]. Based on what is currently known, further studies investigating the role of the PAS in AD and other dementias are certainly warranted.
DECLARATIONS
AcknowledgmentsThe authors gratefully acknowledge Dr. Suzette Smiley-Jewell for assistance with manuscript preparation.
Authors’ contributionsMade substantial contributions to conception and outline of the review: Tang MY, Gorin FA, Lein PJ
Performed literature search to identify relevant publications: Tang MY, Gorin FA
Wrote the initial draft of the manuscript and created figures: Tang MY
Significantly edited early versions of the manuscript: Gorin FA
Made final edits to the manuscript: Lein PJ
Obtained funding to support the work: Gorin FA, Lein PJ
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by the National Institute of Aging (Grant No. P30 AG010129; No. R21 AG065908). The National Institute of Aging (NIA) was not involved in the design, layout, writing or decision to submit this review for publication. The contents of this work do not necessarily represent the official views of the NIA, and the NIA does not endorse the purchase of any commercial products or services mentioned in this publication.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. Morris GP, Clark IA, Vissel B. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol Commun 2014;2:135.
4. Janelidze S, Mattsson N, Palmqvist S, et al. Plasma P-tau181 in Alzheimer’s disease: relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer’s dementia. Nat Med 2020;26:379-86.
5. Honig LS, Vellas B, Woodward M, et al. Trial of Solanezumab for mild dementia due to Alzheimer’s disease. N Engl J Med 2018;378:321-30.
6. Decourt B, Boumelhem F, Pope ED 3rd, Shi J, Mari Z, Sabbagh MN. Critical appraisal of amyloid lowering agents in AD. Curr Neurol Neurosci Rep 2021;21:39.
7. Egan MF, Kost J, Tariot PN, et al. Randomized trial of verubecestat for mild-to-moderate Alzheimer’s disease. N Engl J Med 2018;378:1691-703.
8. Gauthier S, Feldman HH, Schneider LS, et al. Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: a randomised, controlled, double-blind, parallel-arm, phase 3 trial. Lancet 2016;388:2873-84.
11. Nelson PT, Alafuzoff I, Bigio EH, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol 2012;71:362-81.
12. Mehra A, Ali C, Parcq J, Vivien D, Docagne F. The plasminogen activation system in neuroinflammation. Biochim Biophys Acta 2016;1862:395-402.
13. Akiyama H, Barger S, Barnum S, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging 2000;21:383-421.
14. Jevtic S, Sengar AS, Salter MW, McLaurin J. The role of the immune system in Alzheimer disease: etiology and treatment. Ageing Res Rev 2017;40:84-94.
15. Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement (N Y) 2018;4:575-90.
16. Heneka MT, Carson MJ, Khoury JE, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol 2015;14:388-405.
17. Ni J, Wu Z. Inflammation spreading: negative spiral linking systemic inflammatory disorders and Alzheimer’s disease. Front Cell Neurosci 2021;15:638686.
18. Oh S, Suh N, Kim I, Lee JY. Impacts of aging and amyloid-β deposition on plasminogen activators and plasminogen activator inhibitor-1 in the Tg2576 mouse model of Alzheimer’s disease. Brain Res 2015;1597:159-67.
19. Lee JY, Kweon HS, Cho E, et al. Upregulation of tPA/plasminogen proteolytic system in the periphery of amyloid deposits in the Tg2576 mouse model of Alzheimer’s disease. Neurosci Lett 2007;423:82-7.
20. Ries M, Sastre M. Mechanisms of Aβ clearance and degradation by glial cells. Front Aging Neurosci 2016;8:160.
21. Tarasoff-Conway JM, Carare RO, Osorio RS, et al. Clearance systems in the brain-implications for Alzheimer disease. Nat Rev Neurol 2015;11:457-70.
22. Gerrits E, Brouwer N, Kooistra SM, et al. Distinct amyloid-β and tau-associated microglia profiles in Alzheimer’s disease. Acta Neuropathol 2021;141:681-96.
23. Hong S, Dissing-Olesen L, Stevens B. New insights on the role of microglia in synaptic pruning in health and disease. Curr Opin Neurobiol 2016;36:128-34.
24. Rajendran L, Paolicelli RC. Microglia-mediated synapse loss in Alzheimer’s disease. J Neurosci 2018;38:2911-9.
25. Stephan AH, Barres BA, Stevens B. The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci 2012;35:369-89.
26. Liddelow SA, Guttenplan KA, Clarke LE, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017;541:481-7.
28. Olmos-Alonso A, Schetters ST, Sri S, et al. Pharmacological targeting of CSF1R inhibits microglial proliferation and prevents the progression of Alzheimer’s-like pathology. Brain 2016;139:891-907.
29. Hong S, Beja-Glasser VF, Nfonoyim BM, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016;352:712-6.
30. Cisbani G, Rivest S. Targeting innate immunity to protect and cure Alzheimer’s disease: opportunities and pitfalls. Mol Psychiatry 2021;26:5504-15.
31. Heissig B, Salama Y, Takahashi S, Osada T, Hattori K. The multifaceted role of plasminogen in inflammation. Cell Signal 2020;75:109761.
32. Newcombe EA, Camats-Perna J, Silva ML, Valmas N, Huat TJ, Medeiros R. Inflammation: the link between comorbidities, genetics, and Alzheimer’s disease. J Neuroinflammation 2018;15:276.
33. Le Page A, Dupuis G, Frost EH, et al. Role of the peripheral innate immune system in the development of Alzheimer’s disease. Exp Gerontol 2018;107:59-66.
34. Svenningsen P, Hinrichs GR, Zachar R, Ydegaard R, Jensen BL. Physiology and pathophysiology of the plasminogen system in the kidney. Pflugers Arch 2017;469:1415-23.
35. Calderón-Garcidueñas L, Franco-Lira M, Mora-Tiscareño A, Medina-Cortina H, Torres-Jardón R, Kavanaugh M. Early Alzheimer’s and Parkinson’s disease pathology in urban children: Friend versus Foe responses--it is time to face the evidence. Biomed Res Int 2013;2013:161687.
36. Calderón-Garcidueñas L, Kavanaugh M, Block M, et al. Neuroinflammation, hyperphosphorylated tau, diffuse amyloid plaques, and down-regulation of the cellular prion protein in air pollution exposed children and young adults. J Alzheimers Dis 2012;28:93-107.
37. Jialal I, Devaraj S. Subcutaneous adipose tissue biology in metabolic syndrome. Horm Mol Biol Clin Investig 2018:33.
38. Zhang B, Wang HE, Bai YM, et al. Inflammatory bowel disease is associated with higher dementia risk: a nationwide longitudinal study. Gut 2021;70:85-91.
39. Chana-Muñoz A, Jendroszek A, Sønnichsen M, et al. Origin and diversification of the plasminogen activation system among chordates. BMC Evol Biol 2019;19:27.
40. Cesari M, Pahor M, Incalzi RA. Plasminogen activator inhibitor-1 (PAI-1): a key factor linking fibrinolysis and age-related subclinical and clinical conditions. Cardiovasc Ther 2010;28:e72-91.
41. Kruithof EK. Regulation of plasminogen activator inhibitor type 1 gene expression by inflammatory mediators and statins. Thromb Haemost 2008;100:969-75.
42. Lee TW, Tsang VW, Birch NP. Physiological and pathological roles of tissue plasminogen activator and its inhibitor neuroserpin in the nervous system. Front Cell Neurosci 2015;9:396.
43. Ricagno S, Caccia S, Sorrentino G, Antonini G, Bolognesi M. Human neuroserpin: structure and time-dependent inhibition. J Mol Biol 2009;388:109-21.
44. Chandler WL, Alessi MC, Aillaud MF, Henderson P, Vague P, Juhan-Vague I. Clearance of tissue plasminogen activator (TPA) and TPA/plasminogen activator inhibitor type 1 (PAI-1) complex: relationship to elevated TPA antigen in patients with high PAI-1 activity levels. Circulation 1997;96:761-8.
45. Castellino FJ, Ploplis VA. Structure and function of the plasminogen/plasmin system. Thromb Haemost 2005;93:647-54.
46. Andreasen PA, Egelund R, Petersen HH. The plasminogen activation system in tumor growth, invasion, and metastasis. Cell Mol Life Sci 2000;57:25-40.
47. Zhang L, Seiffert D, Fowler BJ, Jenkins GR, Thinnes TC, et al. Plasminogen has a broad extrahepatic distribution. Thromb Haemost 2002;87:493-501.
49. Tjärnlund-Wolf A, Brogren H, Lo EH, Wang X. Plasminogen activator inhibitor-1 and thrombotic cerebrovascular diseases. Stroke 2012;43:2833-9.
50. Atsev S, Tomov N. Using antifibrinolytics to tackle neuroinflammation. Neural Regen Res 2020;15:2203-6.
51. Baker SK, Strickland S. A critical role for plasminogen in inflammation. J Exp Med 2020;217:e20191865.
52. Diaz A, Merino P, Guo JD, et al. Urokinase-type plasminogen activator protects cerebral cortical neurons from soluble Aβ-induced synaptic damage. J Neurosci 2020;40:4251-63.
53. Diaz A, Merino P, Manrique LG, et al. A cross talk between neuronal urokinase-type plasminogen activator (uPA) and astrocytic uPA receptor (uPAR) promotes astrocytic activation and synaptic recovery in the ischemic brain. J Neurosci 2017;37:10310-22.
54. Jeanneret V, Ospina JP, Diaz A, et al. Tissue-type plasminogen activator protects the postsynaptic density in the ischemic brain. J Cereb Blood Flow Metab 2018;38:1896-910.
55. Yepes M. The plasminogen activating system in the pathogenesis of Alzheimer’s disease. Neural Regen Res 2021;16:1973-7.
56. Chevilley A, Lesept F, Lenoir S, Ali C, Parcq J, Vivien D. Impacts of tissue-type plasminogen activator (tPA) on neuronal survival. Front Cell Neurosci 2015;9:415.
57. Lenoir S, Varangot A, Lebouvier L, Galli T, Hommet Y, Vivien D. Post-synaptic release of the neuronal tissue-type plasminogen activator (tPA). Front Cell Neurosci 2019;13:164.
58. Sappino AP, Madani R, Huarte J, et al. Extracellular proteolysis in the adult murine brain. J Clin Invest 1993;92:679-85.
59. Vassalli J, Belin D. Amiloride selectively inhibits the urokinase-type plasminogen activator. FEBS Letters 1987;214:187-91.
60. Yepes M, Lawrence DA. Neuroserpin: a selective inhibitor of tissue-type plasminogen activator in the central nervous system. Thromb Haemost 2004;91:457-64.
61. Lee TW, Tsang VW, Loef EJ, Birch NP. Physiological and pathological functions of neuroserpin: regulation of cellular responses through multiple mechanisms. Semin Cell Dev Biol 2017;62:152-9.
62. Davis RL, Shrimpton AE, Holohan PD, et al. Familial dementia caused by polymerization of mutant neuroserpin. Nature 1999;401:376-9.
63. Salles FJ, Strickland S. Localization and regulation of the tissue plasminogen activator-plasmin system in the hippocampus. J Neurosci 2002;22:2125-34.
64. Stevenson TK, Lawrence DA. Characterization of tissue plasminogen activator expression and trafficking in the adult murine brain. eNeuro 2018;5:ENEURO.
65. Zhu J, Wan Y, Xu H, Wu Y, Hu B, Jin H. The role of endogenous tissue-type plasminogen activator in neuronal survival after ischemic stroke: friend or foe? Cell Mol Life Sci 2019;76:1489-506.
66. Melchor JP, Strickland S. Tissue plasminogen activator in central nervous system physiology and pathology. Thromb Haemost 2005;93:655-60.
67. Seeds NW, Basham ME, Ferguson JE. Absence of tissue plasminogen activator gene or activity impairs mouse cerebellar motor learning. J Neurosci 2003;23:7368-75.
68. Yepes M. The plasminogen activation system promotes neurorepair in the ischemic brain. Curr Drug Targets 2019;20:953-9.
69. Yepes M. Tissue-type plasminogen activator is a neuroprotectant in the central nervous system. Front Cell Neurosci 2015;9:304.
70. Yepes M, Sandkvist M, Moore EG, Bugge TH, Strickland DK, Lawrence DA. Tissue-type plasminogen activator induces opening of the blood-brain barrier via the LDL receptor-related protein. J Clin Invest 2003;112:1533-40.
71. Dent MA, Sumi Y, Morris RJ, Seeley PJ. Urokinase-type plasminogen activator expression by neurons and oligodendrocytes during process outgrowth in developing rat brain. Eur J Neurosci 1993;5:633-47.
72. Merino P, Diaz A, Jeanneret V, et al. Urokinase-type plasminogen activator (uPA) binding to the uPA receptor (uPAR) promotes axonal regeneration in the central nervous system. J Biol Chem 2017;292:2741-53.
73. Jacobsen JS, Comery TA, Martone RL, et al. Enhanced clearance of Abeta in brain by sustaining the plasmin proteolysis cascade. Proc Natl Acad Sci U S A 2008;105:8754-9.
74. Kutz SM, Higgins CE, Higgins PJ. Novel combinatorial therapeutic targeting of PAI-1 (SERPINE1) gene expression in Alzheimer’s disease. Mol Med Ther 2012;1:106.
75. Ledesma MD, Da Silva JS, Crassaerts K, Delacourte A, De Strooper B, Dotti CG. Brain plasmin enhances APP alpha-cleavage and Abeta degradation and is reduced in Alzheimer’s disease brains. EMBO Rep 2000;1:530-5.
76. Liu RM, van Groen T, Katre A, et al. Knockout of plasminogen activator inhibitor 1 gene reduces amyloid beta peptide burden in a mouse model of Alzheimer’s disease. Neurobiol Aging 2011;32:1079-89.
77. Tucker HM, Kihiko M, Caldwell JN, et al. The plasmin system is induced by and degrades amyloid-beta aggregates. J Neurosci 2000;20:3937-46.
78. Oh SB, Byun CJ, Yun JH, et al. Tissue plasminogen activator arrests Alzheimer’s disease pathogenesis. Neurobiol Aging 2014;35:511-9.
79. Melchor JP, Pawlak R, Strickland S. The tissue plasminogen activator-plasminogen proteolytic cascade accelerates amyloid-beta (Abeta) degradation and inhibits Abeta-induced neurodegeneration. J Neurosci 2003;23:8867-71.
80. Tucker HM, Kihiko-Ehmann M, Estus S. Urokinase-type plasminogen activator inhibits amyloid-beta neurotoxicity and fibrillogenesis via plasminogen. J Neurosci Res 2002;70:249-55.
81. Tucker HM, Kihiko-Ehmann M, Wright S, Rydel RE, Estus S. Tissue plasminogen activator requires plasminogen to modulate amyloid-beta neurotoxicity and deposition. J Neurochem 2000;75:2172-7.
82. Samarakoon R, Higgins SP, Higgins CE, Higgins PJ. The TGF-β1/p53/PAI-1 signaling axis in vascular senescence: role of caveolin-1. Biomolecules 2019;9:341.
83. Vaughan DE, Rai R, Khan SS, Eren M, Ghosh AK. Plasminogen activator inhibitor-1 is a marker and a mediator of senescence. Arterioscler Thromb Vasc Biol 2017;37:1446-52.
84. Yamamoto K, Takeshita K, Kojima T, Takamatsu J, Saito H. Aging and plasminogen activator inhibitor-1 (PAI-1) regulation: implication in the pathogenesis of thrombotic disorders in the elderly. Cardiovasc Res 2005;66:276-85.
85. Oh J, Lee HJ, Song JH, Park SI, Kim H. Plasminogen activator inhibitor-1 as an early potential diagnostic marker for Alzheimer’s disease. Exp Gerontol 2014;60:87-91.
86. Akhter H, Huang WT, van Groen T, Kuo HC, Miyata T, Liu RM. A small molecule inhibitor of plasminogen activator inhibitor-1 reduces brain amyloid-β load and improves memory in an animal model of Alzheimer’s disease. J Alzheimers Dis 2018;64:447-57.
87. Hohensinner PJ, Baumgartner J, Kral-Pointner JB, et al. PAI-1 (plasminogen activator inhibitor-1) expression renders alternatively activated human macrophages proteolytically quiescent. Arterioscler Thromb Vasc Biol 2017;37:1913-22.
88. Medina MG, Ledesma MD, Domínguez JE, et al. Tissue plasminogen activator mediates amyloid-induced neurotoxicity via Erk1/2 activation. EMBO J 2005;24:1706-16.
89. Pineda D, Ampurdanés C, Medina MG, et al. Tissue plasminogen activator induces microglial inflammation via a noncatalytic molecular mechanism involving activation of mitogen-activated protein kinases and Akt signaling pathways and AnnexinA2 and Galectin-1 receptors. Glia 2012;60:526-40.
90. Hou B, Eren M, Painter CA, et al. Tumor necrosis factor alpha activates the human plasminogen activator inhibitor-1 gene through a distal nuclear factor kappaB site. J Biol Chem 2004;279:18127-36.
91. Loures CMG, Duarte RCF, Silva MVF, et al. Hemostatic abnormalities in dementia: a systematic review and meta-analysis. Semin Thromb Hemost 2019;45:514-22.
92. Sutton R, Keohane ME, VanderBerg SR, Gonias SL. Plasminogen activator inhibitor-1 in the cerebrospinal fluid as an index of neurological disease. Blood Coagul Fibrinolysis 1994;5:167-71.
93. Barker R, Kehoe PG, Love S. Activators and inhibitors of the plasminogen system in Alzheimer’s disease. J Cell Mol Med 2012;16:865-76.
94. Higgins PJ. The TGF-beta1/upstream stimulatory factor-regulated PAI-1 gene: potential involvement and a therapeutic target in Alzheimer’s disease. J Biomed Biotechnol 2006;2006:15792.
95. Kawarada Y, Inoue Y, Kawasaki F, et al. TGF-β induces p53/Smads complex formation in the PAI-1 promoter to activate transcription. Sci Rep 2016;6:35483.
96. Jeon H, Kim JH, Kim JH, Lee WH, Lee MS, Suk K. Plasminogen activator inhibitor type 1 regulates microglial motility and phagocytic activity. J Neuroinflammation 2012;9:149.
97. Cao C, Lawrence DA, Li Y, et al. Endocytic receptor LRP together with tPA and PAI-1 coordinates Mac-1-dependent macrophage migration. EMBO J 2006;25:1860-70.
98. Hamilton JA, Whitty GA, Wojta J, Gallichio M, McGrath K, Ianches G. Regulation of plasminogen activator inhibitor-1 levels in human monocytes. Cell Immunol 1993;152:7-17.
99. Fabbro S, Seeds NW. Plasminogen activator activity is inhibited while neuroserpin is up-regulated in the Alzheimer disease brain. J Neurochem 2009;109:303-15.
100. Angelucci F, Čechová K, Průša R, Hort J. Amyloid beta soluble forms and plasminogen activation system in Alzheimer’s disease: Consequences on extracellular maturation of brain-derived neurotrophic factor and therapeutic implications. CNS Neurosci Ther 2019;25:303-13.
101. Mutimer CA, Keragala CB, Markus HS, Werring DJ, Cloud GC, Medcalf RL. Cerebral amyloid angiopathy and the fibrinolytic system: is plasmin a therapeutic target? Stroke 2021;52:2707-14.
102. Park L, Zhou J, Koizumi K, et al. tPA deficiency underlies neurovascular coupling dysfunction by amyloid-β. J Neurosci 2020;40:8160-73.
103. Deane R, Zlokovic BV. Role of the blood-brain barrier in the pathogenesis of Alzheimer’s disease. Curr Alzheimer Res 2007;4:191-7.
104. Zenaro E, Piacentino G, Constantin G. The blood-brain barrier in Alzheimer’s disease. Neurobiol Dis 2017;107:41-56.
105. Huang X, Hussain B, Chang J. Peripheral inflammation and blood-brain barrier disruption: effects and mechanisms. CNS Neurosci Ther 2021;27:36-47.
106. Bardehle S, Rafalski VA, Akassoglou K. Breaking boundaries-coagulation and fibrinolysis at the neurovascular interface. Front Cell Neurosci 2015;9:354.
107. Cortes-Canteli M, Mattei L, Richards AT, Norris EH, Strickland S. Fibrin deposited in the Alzheimer’s disease brain promotes neuronal degeneration. Neurobiol Aging 2015;36:608-17.
108. Ryu JK, Rafalski VA, Meyer-Franke A, et al. Fibrin-targeting immunotherapy protects against neuroinflammation and neurodegeneration. Nat Immunol 2018;19:1212-23.
109. Kim JS. tPA helpers in the treatment of acute ischemic stroke: are they ready for clinical use? J Stroke 2019;21:160-74.
110. Lopes Pinheiro MA, Kooij G, Mizee MR, et al. Immune cell trafficking across the barriers of the central nervous system in multiple sclerosis and stroke. Biochim Biophys Acta 2016;1862:461-71.
111. Sarvari S, Moakedi F, Hone E, Simpkins JW, Ren X. Mechanisms in blood-brain barrier opening and metabolism-challenged cerebrovascular ischemia with emphasis on ischemic stroke. Metab Brain Dis 2020;35:851-68.
112. Spuch C, Ortolano S, Navarro C. LRP-1 and LRP-2 receptors function in the membrane neuron. Trafficking mechanisms and proteolytic processing in Alzheimer’s disease. Front Physiol 2012;3:269.
113. Cai Z, Qiao PF, Wan CQ, Cai M, Zhou NK, Li Q. Role of blood-brain barrier in Alzheimer’s disease. J Alzheimers Dis 2018;63:1223-34.
114. Lewandowski SA, Fredriksson L, Lawrence DA, Eriksson U. Pharmacological targeting of the PDGF-CC signaling pathway for blood-brain barrier restoration in neurological disorders. Pharmacol Ther 2016;167:108-19.
115. Su EJ, Cao C, Fredriksson L, et al. Microglial-mediated PDGF-CC activation increases cerebrovascular permeability during ischemic stroke. Acta Neuropathol 2017;134:585-604.
116. Benchenane K, Berezowski V, Ali C, et al. Tissue-type plasminogen activator crosses the intact blood-brain barrier by low-density lipoprotein receptor-related protein-mediated transcytosis. Circulation 2005;111:2241-9.
117. Abu Fanne R, Nassar T, Yarovoi S, et al. Blood-brain barrier permeability and tPA-mediated neurotoxicity. Neuropharmacology 2010;58:972-80.
118. Kaur J, Tuor UI, Zhao Z, Barber PA. Quantitative MRI reveals the elderly ischemic brain is susceptible to increased early blood-brain barrier permeability following tissue plasminogen activator related to claudin 5 and occludin disassembly. J Cereb Blood Flow Metab 2011;31:1874-85.
119. Golias C, Charalabopoulos A, Stagikas D, Charalabopoulos K, Batistatou A. The kinin system--bradykinin: biological effects and clinical implications. Multiple role of the kinin system--bradykinin. Hippokratia 2007;11:124-8.
120. Marcos-Contreras OA, Martinez de Lizarrondo S, Bardou I, et al. Hyperfibrinolysis increases blood-brain barrier permeability by a plasmin- and bradykinin-dependent mechanism. Blood 2016;128:2423-34.
121. Gauberti M, Potzeha F, Vivien D, Martinez de Lizarrondo S. Impact of bradykinin generation during thrombolysis in ischemic stroke. Front Med (Lausanne) 2018;5:195.
122. Simão F, Feener EP. The effects of the contact activation system on hemorrhage. Front Med (Lausanne) 2017;4:121.
123. Calderón-Garcidueñas L, Reynoso-Robles R, Vargas-Martínez J, et al. Prefrontal white matter pathology in air pollution exposed Mexico City young urbanites and their potential impact on neurovascular unit dysfunction and the development of Alzheimer’s disease. Environ Res 2016;146:404-17.
124. Ji HL, Zhao R, Matalon S, Matthay MA. Elevated plasmin(ogen) as a common risk factor for COVID-19 susceptibility. Physiol Rev 2020;100:1065-75.
125. Nation DA, Sweeney MD, Montagne A, et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med 2019;25:270-6.
126. Sweeney MD, Sagare AP, Zlokovic BV. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol 2018;14:133-50.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Tang MY, Gorin FA, Lein PJ. Review of evidence implicating the plasminogen activator system in blood-brain barrier dysfunction associated with Alzheimer's disease. Ageing Neur Dis 2022;2:2. http://dx.doi.org/10.20517/and.2022.05
AMA Style
Tang MY, Gorin FA, Lein PJ. Review of evidence implicating the plasminogen activator system in blood-brain barrier dysfunction associated with Alzheimer's disease. Ageing and Neurodegenerative Diseases. 2022; 2(1): 2. http://dx.doi.org/10.20517/and.2022.05
Chicago/Turabian Style
Tang, Mei-Yun, Fredric A. Gorin, Pamela J. Lein. 2022. "Review of evidence implicating the plasminogen activator system in blood-brain barrier dysfunction associated with Alzheimer's disease" Ageing and Neurodegenerative Diseases. 2, no.1: 2. http://dx.doi.org/10.20517/and.2022.05
ACS Style
Tang, M.Y.; Gorin FA.; Lein PJ. Review of evidence implicating the plasminogen activator system in blood-brain barrier dysfunction associated with Alzheimer's disease. Ageing. Neur. Dis. 2022, 2, 2. http://dx.doi.org/10.20517/and.2022.05
About This Article
Copyright

Data & Comments
Data


 Cite This Article 27 clicks
Cite This Article 27 clicks

 Like This Article 10
likes
Like This Article 10
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.