
Naive BM-derived stem cells (Neuro-Cells) may modify acute and chronic neurodegenerative disorders by modulating macrophage behaviors

 , ...
, ... Abstract
In acute traumatic or hypoxic brain and spinal cord lesions, as well as in chronic idiopathic neurodegenerative disorders induced by a genetic/environmental/idiopathic protein misfolding with aggregation, emerging evidence indicates that primary necrosis, as induced by the underlying event, initiates a secondary inflammatory process. In this secondary process, responsible for significant neurological deterioration, a microglia type M1/M2 misbalance plays a major role. Indeed, both acute and chronic neurodegenerative disorders share a common pathway: a M1/M2 misbalance-induced hyperinflammatory process with a lack of response to conventional anti-inflammatory interventions. In recent literature, however, both in preclinical and clinical neurodegenerative conditions, these processes were suggested to be sensitive for interventions with stem cells. Intrathecal interventions with a fresh, not-manipulated (naïve) bone marrow-derived stem cell preparation, after positive selection of pro-inflammatory substances (Neuro-Cells), were found to prevent/reduce secondary necrosis-induced pro-inflammatory and pro-apoptotic processes in both immune-compromised and otherwise healthy experimental animal models. Therefore, it seems justified to further encourage clinical trials applying autologous BM-derived naïve stem cells in patients suffering from those debilitating neurodegenerative conditions.
Keywords
INTRODUCTION
In any acute or chronic, systemic or compartmental insults, interferons produced by lesioned cells are responsible for a range of signaling events leading to an inflammatory process, eventually ending with apoptosis and/or necrosis. Chemokines will activate immune cells such as macrophages and microglia to travel to the site of the insult. Exposed to inflammatory stimuli, these cells will initiate the secretion of cytokines.
Adequate resolution of the inflammatory process with phagocytosis of cell debris, cell survival and tissue repair, will be reached when the temporal dynamics of these cytokines show an early innate immune response with a release of pro-inflammatory tumor necrosis factor (TNF)-α, followed by a release of interferon (IFN)-γ, and then mainly interleukin (IL)-1β, IL-6, and IL-12. Normally, an adequate number of anti-inflammatory cytokines such as IL-4 and IL-10 will be subsequently released in response to the prior pro-inflammatory cytokines. However, an inadequate counter-balancing level of anti-inflammatory cytokines, or an overzealous/prolonged secretion of pro-inflammatory cytokines might cause a vicious, hyperinflammatory cycle [Figure 1]. Increasing inappropriate cytokine release-related morbidity includes multi-organ failure, neurotoxicity, and death[1-2].
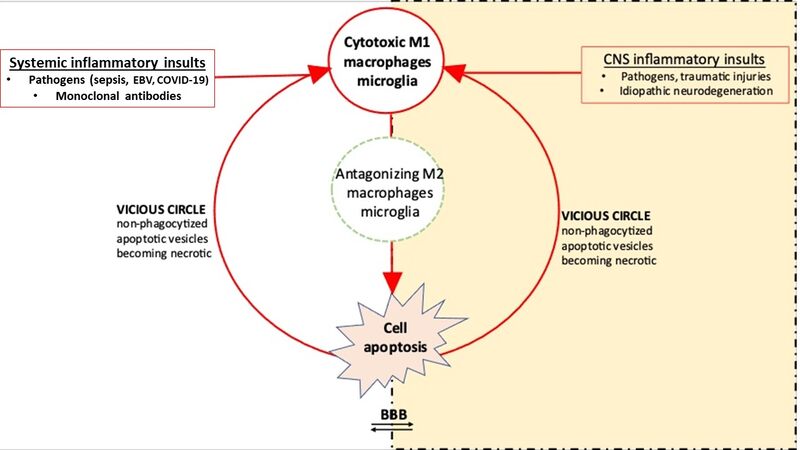
Figure 1. The origin and propagation (vicious cycle) of hyperinflammation. In case of hyperinflammation, inappropriate (increased) levels of pro-inflammatory cytokines or (decreased) levels of anti-inflammatory cytokines (M1/M2 paradigm) initiate a vicious circle as non-phagocytized apoptotic vesicles become necrotic and continue the activation of resting macrophages/microglia into M1 macrophages/microglia. Thus, ongoing signaling pathways prolong the cytokine cascade with activation of other immune cell types which promote cell proliferation, boost the pro-inflammatory cytokine release (mainly IL-6), and thus promotes the propagation of tissue damage. In the figure, the blood-brain-barrier (BBB) is shown with the systemic compartment (blood) on the left and the CNS compartment at the right side. In case of hyperinflammation, BBB permeability is increased.
Indeed, a systemic or compartmental disbalance between pro- and anti-inflammatory cytokines (i.e., a disturbed microglia type M1/M2 balance) may result in hyperinflammatory conditions and/or cytokine release syndromes (CRS). These conditions might be best formulated as an infectious or otherwise-induced production of circulating cytokines beyond a normal response, leading to inflammatory signs with fever, severe fatigue, nausea, and in some cases even secondary organ dysfunction or multi-organ failure[3]. Infectious insults include sepsis, viremia, herpes, Ebola, malaria, Dengue, Lassa, and coronavirus-induced severe acute respiratory syndrome or Middle East respiratory syndrome. Sterile conditions, such as monogenic disorders, autoimmune diseases, organ transplantation, immunotherapies like monoclonal antibodies or chimere antigen receptor-T cells for cancer, as well as burns, ischemia, and trauma, may also initiate inappropriate cytokine secretion[4-6]. The insults may be acute, subacute, or chronic. In chronic neurodegenerative disorders, chronic misfolding of proteins with subsequent divergent accumulation and aggregates formation as well as ongoing cell necrosis can be related to a disturbed M1/M2 paradigm with an elevation in the M1 pro‐inflammatory phenotype by the continuous exposure to pathogen-associated molecular patterns (PAMPs) and/or endogenous damage-associated molecular patterns (DAMPs). A similar shift towards M1 polarization might also be seen in rheumatoid arthritis arthritis[7].
CRS manifest with fever and general malaise, but as soon as endothelial cells become involved it may also come with coagulopathy, capillary leaks, and disruption of membranes. Also membranes surrounding immune-privileged compartments such as the blood-brain-barrier might be affected and lose their relative impermeability, thus enabling immune cells to freely pass those membranes[8]. The massive intracerebral influx of macrophages due to this increased permeability of the blood-brain-barrier explains the progressive exacerbation in chronic progressive neurodegenerative disorders. If untreated, most patients will suffer a diffuse intravascular coagulation and/or a pro-thrombotic coagulopathy with thrombocytopenia, leading to hypotension, multi-organ failure and/or acute hypoxemia[1].
In the absence of external stimuli, macrophages and microglia are normally in a resting state (M0 macrophages/microglia). Due to polarization, macrophages adopt different functional programs in response to microenvironmental signals [Figure 2]. Pending their micro-environment and the presence of polarizing cytokines, they may be classically activated into M1 phenotypes, as well as into alternatively activated into M2 phenotypes. In the M1 state, macrophages/microglia secrete pro-inflammatory responses, enhancing nitric oxide synthase. In the M2 state, in addition to stimulating responses for repair and recruitment (from M2a and M2b phenotypes, respectively) they also may secrete anti-inflammatory phagocytic responses (M2c phenotypes)[9]. Indeed, macrophages also play an important role in the embryonic development, removal of cellular debris, and tissue repair. The polarization of mononuclear macrophages into M1 or M2 macrophages is a simplified conceptual framework to describe their plasticity[10]. Originally, macrophages were thought to be activated by IFN-γ alone or in concert with microbial stimuli (e.g., lipopolysaccharide) or cytokines (e.g., granulocyte-macrophage colony-stimulating factor) (so called classically activated macrophages). Subsequently, macrophage colony stimulating factor, TNF-β and the interleukins IL-4 and IL-10, rather than inhibiting this classical activation, were found to induce an alternative (M2) form of macrophage activation [Figure 2]. In response to certain endogenous and exogenous conditions, macrophages may even reverse classical or alternative polarization.
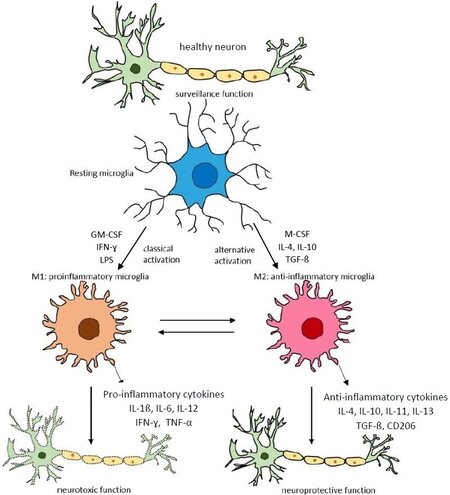
Figure 2. The M1/M2 paradigm in the central nervous system. Transcriptional regulators of M1 and M2 activation of microglia and mechanisms of their stimulation/inhibition. Resting microglia are stimulated by interferon (IFN)-γ, lipopolysaccharide (LPS), and/or granulocyte-macrophage colony-stimulating factor (GM-CSF) to classical activation into M1 microglia, and by macrophage colony-stimulating factor (M-CSF), IL-4, IL-10, and transforming growth factor (TGF)-β for alternative activation into M2 microglia, producing pro-inflammatory and anti-inflammatory cytokines, respectively. In a well-balanced M1/M2 condition, there will be an adequate resolution of the inflammatory process. TNF-α: Tumour necrosis factor α; CD206: macrophage mannose receptor type 1 (adapted from
Although different patterns of macrophage responses cannot always be accurately described along the M1/M2 axis (in some reactive microglial populations, the canonical gene products of both “polarized” states are co-expressed)[11], the M1/M2 axis simply reflects the most phenotypically polar differentiation states of macrophages and, therefore, is often implied in research. In in vivo studies, the M1/M2 dichotomy may possibly be replaced with the terms pro-inflammatory/pro-regenerative[12].
In the acute phase of an insult, M1 macrophages phagocytose the debris and promote the flow of other immune cells by expressing pro-inflammatory cytokines, mainly IL-1β, IL-6, IL-12, TNF-α, and IFN-γ [Figure 2].
After the acute phase with the onset of classical pro-inflammatory activation of the resting homeostatic M0 macrophages/microglia into M1 phenotypes, normally within 3-7 days later, the transition to regeneration is reflected by increasing numbers of alternatively activated (M2) macrophages, dampening the pro-inflammatory M1 cells-induced immune responses, and promoting regeneration and angiogenesis by expressing anti-inflammatory cytokines such as tumor growth factor (TGF)-β and the interleukins IL-4,
In the recent past, more attention is given to the role of neuro-inflammation as a common final pathway in neurodegenerative disorders. Indeed, neuro-inflammation (i.e., gliosis and inflammatory reactions) has been described as a prominent sign in Alzheimer’s disease[10,13], Parkinson’s disease[14], Huntington disease[15], amyotrophic lateral sclerosis[16], prion disease[17], and multiple sclerosis[18,19]. In these disorders, chronic protein misfolding maintains a disturbed M1/M2 paradigm by the continuous exposure to PAMPs and/or DAMPs.
INAPPROPRIATE CYTOKINE RELEASE SYNDROMES
Macrophages and microglia are the sentries of the innate immune system in injury and infection; they are thought to play a major role in the tissue and organ homeostasis, as well as in autoimmune diseases, atherosclerosis, and cancer.
The timely switching of macrophage polarization from M1 to M2 plays a major role in the outcome of the inflammatory reaction (regeneration or fibrosis). Adequate immunosuppression and neuron protection is pending from a normal M1/M2 paradigm[10]. In case of a disturbed paradigm, a necrotic cell-induced hyper-inflammatory condition may result, due to a vicious circle with an ongoing classical activation of the microglia, a condition with a great deal of collateral damage[20-22] [Figures 1 and 3].
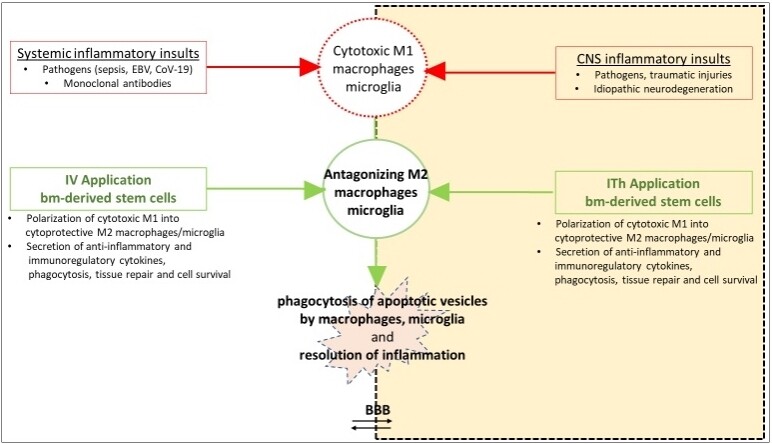
Figure 3. The breaking of the vicious cycle in hyperinflammatory conditions by bone marrow-derived naive stem cells. Necrosis in the target tissues initiate an M1 macrophage/microglia-induced cytokine cascade and activation of other cell types resulting in cell proliferation, further pro-inflammatory cytokine release (mainly IL-6), and propagation of tissue damage. In case of an imbalanced release of pro- and anti-inflammatory cytokines, phagocytosis of the apoptotic vesicles is blocked, and a vicious cycle will follow by the non-phagocytized vesicles, becoming necrotic. Intravenous (IV) and/or intrathecal (ITh) transplantation of stem cells then may restore the imbalanced cytokine levels and break the vicious cycle by polarization of cytotoxic M1 into antagonizing M2 immune cells on the one, and inhibition of the up‐regulation of the protein expression of inflammatory markers (GSK-3β) on the other hand.
Both in systemic and CNS (central nervous system) compartmental inappropriate CRS, the blood-brain-barrier permeability normally preventing for the infiltration of blood‐borne monocytes/macrophages may be compromised, allowing the passage of chemokines, immune cells, and cytokines[8,23-28]. Thus, systemic hyper-inflammation may manifest with encephalopathic manifestations and a permanent deterioration in pre-existing neurodegenerative disorders[2,29,30].
In recent years, in several neurodegenerative diseases, the M1/M2 paradigm of microglial activation was extensively studied to uncover the mechanisms of immunopathogenesis. Molecular and clinical evidence from positron emission tomography imaging and post-mortem analysis suggested an increase of microglial activation and inflammatory mediators during the pathogenesis in these disorders[10]. Predicting the presence and severity of CRS has also been a challenge because this syndrome starts in the target tissue(s), only coming to attention when damage has occurred[1,8]. CRS is a dynamic process and body fluid cytokine levels may not adequately reflect the actual underlying physiological processes[31]. Nevertheless, peripheral blood biomarkers of CRS, even reflecting the remaining situation after cell redistribution to tissues or cell death, are used for diagnosis and to guide therapy[32].
INAPPROPRIATE CYTOKINE SECRETION IN NEURODEGENERATIVE DISORDERS
Neurodegenerative disorders are hereditary and/or sporadic, acute and/or chronic conditions, characterized by nerve cell degeneration and/or necrosis due to atrophy of the nervous system, interfering with normal mental and motor functioning.
Alzheimer’s disease
Alzheimer’s disease (AD) is an age-related multifactorial genetic/environmental neurodegenerative disorder resulting in a progressive impairment in memory, judgement, decision-making, and orientation. In this disorder, intracellular, misfolded tau protein-containing tangles underlie the neurofibrillary degeneration. Microglial macrophages react to the amyloid β peptide by releasing pro-inflammatory factors, promoting their own phagocytic activity[33]. In the immediate vicinity of the characteristic amyloid peptide deposits and neurofibrillary tangles, primarily pro-inflammatory (IL-1ß, IL-6, IL-12, and TNFα) and anti-inflammatory cytokines (IL-4, IL-10, and TGF-β) were found to play a major role in the phagocytic clearance of apoptotic neurons, indicating that inflammation, indeed, is a key pathological hallmark of AD. In Alzheimer’s disease, tau phosphorylation is thought to be responsible for the M1-activated microglia-induced neurotoxicity ease[33-36].
Parkinson’s disease
Parkinson’s disease (PD) is also an age-related genetic/environmental disorder. It is clinically characterized by hypokinesia, bradykinesia, rigidity, and tremor as well as numerous autonomic and mental symptoms, evidencing a multisystem α-synucleinopathic neurodegenerative process. The abundant synuclein characteristically aggregates in Lewy bodies. Both direct and indirect microglial activation are initiated by aggregated α-synuclein. Numerous studies have shown that α-synuclein, probably by its dysregulation of the JAK/STAT pathways in myeloid cells[37], directly activates microglia into the M1 phenotype, with the activation of NADPH oxidase, and increasing production of reactive oxygen species and pro-inflammatory cytokines[38]. In PD patients, increased levels of immune cells and proteins such as adhesion molecules, chemokines, cytokines, and decreased levels of neurotrophins in brain, spinal fluid, and serum, such as brain-derived neurotrophic factor and nerve growth factor, evidenced chronic cytotoxic classical microglial activation with apoptotic cell death[39-43].
Huntington’s disease
Huntington’s disease (HD) is a progressive autosomal dominant monogenic disease, displaying a selective striatal and cortical neuronal loss, manifesting with a progressive motor dysfunction, cognitive decline, and psychiatric disorders. HD is caused by CAG trinucleotide repeat expansion in the gene encoding for huntingtin protein on chromosome 4p16[44]. In HD, proteomic plasma profiling demonstrated that increasing cytokine levels antedate the onset of neurological symptoms. Both in HD patients and experimental animal models, CNS microglial activation was found to result in an increased production of inflammatory mediators, and TNF-α and IL-6 mRNA levels were found markedly increased[45,46].
Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is a mainly sporadic (about 5%-20% familial) multifactorial disease caused by motoneuron degeneration in the spinal cord, brain stem, and primary motor cortex, with cytoplasmic inclusions containing aggregated/ubiquitinated proteins as well as RNAs. In this disease, again, glial activation leads to changes in the expression of a wide range of genes related to the production of soluble molecules, such as cytokines, chemokines, DAMPs, and reactive nitrogen and oxygen species, giving rise to profound modifications in their interactions with neurons[47].
Multiple sclerosis
Multiple sclerosis (MS) is the most common genetic/environmental chronic inflammatory disorder of the CNS, which may manifest as a relapsing-remitting or a secondary progressive disorder[19]. The infiltration of increased autoreactive myelin-specific CD4 and CD8 T helper cells into the CNS represents the crucial event in the inflammatory processes with the formation of focal inflammatory demyelinated lesions (plaques) via the secretion of M1-produced IFN-γ and IFN-γ-promoted TNFα[48-50].
Spinocerebral injuries (SCI) display evidence indicating that immediately after the trauma, macrophages accumulate within the epicenter of the lesion and may initiate necrosis-induced secondary M1 promoted inflammatory mechanisms, overwhelming a comparatively smaller and transient M2 macrophage response, leading to cavities and scar tissue. In time, the acutely increased levels of TNF-α, IL-1β, IFN-γ, and other pro-inflammatory M1-produced cytokines, chemokines, and proteases will gradually decrease, and increasing numbers of anti-inflammatory M2 cytokines will restore the initial M1/M2 balance[51]. Normally, in vitro, myelin phagocytosis comes with a facilitation of M2 polarization; macrophages in a damaged spinal cord are strongly inclined towards M1 polarization, which interferes with the neural tissue recovery.
STEM CELLS
Stem cells are essential for the development, assembling, and repairing of bodily structures. Without these cells one cannot survive. Recently, these cells emerged as a promising tool for the modulation of the immune system. They are undifferentiated cells that not only may proliferate, but also are able to differentiate into all kinds of target cells. Stem cells can be harvested out of adipose tissue, bone marrow, olfactory mucosa, umbilical cord blood, and embryonic tissue, as well as out of special niches in organs, all varying in their regenerative capacity and potency. Their specific differences in biological properties might be an important consideration for their selection in regenerative medicine[52]. Applying autologous stem cells is preferred over allogeneic preparations, as these come with a risk of immunological incompatibility. In regenerative medicine, mostly bone marrow-derived stem cells are applied. Another source of stem cells is supplied by reprogramming adult somatic cells back into pluripotent stem cells: induced pluripotent stem cells. To reach quantitative numbers of stem cells, culturing these cells might help, though this procedure may come with changes in the telomers. Compared to small molecules such as neurotransmitters, and biologics such as antibodies, growth factors, and/or cytokines, stem cells act fundamentally different. However, the exact mechanisms of action of stem cells remain to be elucidated.
The nervous system, unlike many other tissues, has a limited capacity for self-repair; mature nerve cells lack the ability to regenerate, and only neuronal-resident stem cells have the potency to generate new functional neurons in response to lesions. Their limited availability, though, makes them unfit to cure devastating neurodegenerative diseases. Circumvention of this problem through intracerebral neuronal-resident stem cells grafts, on the other hand, raises serious concerns since the pathological phenotype of the diseased endogenous cells may affect the graft tissue. Neuronal-resident stem cells are already predestinated for neuronal renewal-committed operations, whereas naïve stem cells (with an excellent self-renewal capacity with sustained multipotency) are still multi-potent and also exert, for instance, immune-suppressive effects as a dedicated reaction to environmental vesicles or cytokines from degenerating, malfunctioning cells[53].
As said before, originally, the mode of action of bone marrow-derived stem cells was thought to be related to cellular integration by leveraging the plasticity of the stromal/stem and progenitor cells for the replacement of lost cells. Later, the mechanism was also considered to relate indirectly via cellular interactions. As stem cells hardly pass the intact brain barriers, eventual immunosuppressive paracrine and endocrine effects of stem cells in neurodegenerative conditions are rather reached through cell-to-cell interactions by communicators, signaling proteins such as extracellular vesicles, cytokines, growth factors, and/or mitochondrial transfers. Stem cell extracellular vesicles, indeed, were found to exert immune-suppressive effects as a dedicated reaction to environmental vesicles or cytokines from degenerating, malfunctioning cells, thus coordinating their operations with their immediate environment[54-56]. They might be seen as decision making cells. For example, a high concentration of interferon-γ can activate the naïve stem cells to inhibit the innate immune responses, whereas a low concentration will result in the reversed effect.
Autologous stem cell transplants were found to modulate the immune system in both acute[57-59], and chronic[60,61] preclinical and clinical neurodegenerative conditions.
To assure that those stem cells can adapt to local circumstances, it is crucial not to change the multi-potent characteristics of these cells before the cells are re-implanted in the patient. Stem cells have a variety of receptors on their surface, which can be activated by specific antibodies, each changing the polarization of the cell and thus its naïve status[62]. In order to apply naïve stem cells into the environment where neuroinflammation and degeneration are ongoing, in our experiments, fresh human bone marrow-derived stem cells specimen with negatively selected stem cells were manufactured after positive depletion of erythrocytes, monocytes, and lymphocytes, and reduced in volume for intrathecal application (Neuro-Cells: patent WO2015/059300A1). Indeed, intravenous application will end up with most stem cells stuck in lung and liver, and the number of engrafted stem cells reaching the central nervous system will be minimal. Neuro-Cells, intrathecally applied, appeared to be a safe and effective treatment in preclinical models of neurodegeneration as well as in patients. The number of fresh bone marrow-derived stem cells (100 mL bone marrow contains about 108 CD34+ cells) is limited, though, and their half-life is about 72 h. In cases with reduced plasticity of stem cells (e.g., diabetes, renal failure, aging, and severe amyotrophic lateral sclerosis), one may thus consider applying allogenic cells.
As the effects of the stem cells are thought to be reached by cell-to-cell reactions, not the dose but rather the timing is key, as an effective treatment window in acute neurodegenerative processes lies between 24 to 72 h after the initial CNS insult. Similarly, in chronic conditions with ongoing necrosis of neural cells, the best strategy appears to be starting treatment as early as possible, assuming that dead neurons cannot be replaced with this therapy. Here, the key is the slowing down of the ongoing and self-reinforcing disease process by applying the stem cells as early as possible.
STEM CELLS IN THE TREATMENT OF NEURODEGENERATIVE DISORDERS
Regarding systemic CRS, apart from specific vaccines and maybe the anti-viral remdesivir and/or dexamethasone for treatment of some virus-induced syndromes, there are no convincing disease-modifying interventions for those conditions, and symptomatic treatments are still enigmatic. Also, in the treatment of compartmental release syndromes such as in neurodegenerative disorders, due to ambiguous effects and/or serious adverse events, interventions with anti-inflammatory (non)-steroidal anti-inflammatory drugs [(N)SAIDs] were not very successful[63].
As most of acute and chronic, systemic and compartmental CRS, irrespective of their cause, share a common pathophysiological pathway [Figure 1], it seems justified to treat those conditions, in the same way, regardless the phase of the immunological response[2]. Here, adequate understanding of the role of chemokines and cytokines is important for better understanding these syndromes, as well as for diagnostic purposes and the development of therapeutic options. In modern biomedicine, as of now, regulation of cell homeostasis by modulating macrophage behavior in different pathological conditions is key. The M1/M2 paradigm allows the reassessment of the course of typical pathological processes in terms of a misbalanced M1 and M2 macrophage polarization. Here, increasing the relatively low level of M2 macrophage/microglia phenotypes, for instance, might further stimulate regeneration, angiogenesis, and extracellular matrix remodeling. So, in CRS, restoring the M1/M2 phenotype balance might thus lead to restoration of homeostasis and improved clinical symptoms[64]. As pro-inflammatory macrophages are abnormally overrepresented in acute and chronic neurodegenerative disorders, in the next future molecular interventions affecting the M2 subpopulation, therefore, may offer a potential efficient therapeutic approach to suppress or boost the expression of certain genes in these conditions in order to obtain stably polarized M1 or M2 species[49]. The eventual incorporation of cytokines into therapeutic regimens, though, has significant challenges. In addition to low response rates when administered as recombinant proteins and short half-life limiting exposure and efficacy, cytokines can also activate counterregulatory pathways (i.e., immune-potentiating cytokines might initiate immune suppression), thus limiting their potential efficacy[65].
Recent approaches with stem cell implants yielded promising results in patients suffering acute[57,66] and chronic[60,67,69] neurodegenerative disorders. Our own preclinical studies in animal models of acute traumatic spinal cord injury[59,69] and chronic neurodegenerative processes such as amyotrophic lateral sclerosis and frontotemporal lobe degeneration[58,61] were fully in line with these findings. In these experiments, intrathecal application of Neuro-Cells in the various experimental animal models were found to break the hyper-inflammatory process by restoring the normal M1/M2 paradigm [Figure 3].
Those stem cells, but not (N)SAIDs, significantly improved the functional outcome and reduced signs and symptoms of inflammation in these animal models, compared to those treated with placebo, and were free of adverse events. Intrathecal application of Neuro-Cells in SCI-rats within 24 h after the lesioning, induced depolarization of M1 into M2 reactivated macrophages/microglia, thus preventing for the secondary inflammation-induced elevations in serum IL-1β, TNF-α, and IL-6 levels as well as for the elevation of glycogen synthase kinase (GSK)-3β and ionized calcium-binding adaptor molecule (Iba)-1 protein levels in the spinal cord. Those stem cells were found to reduce the SCI-induced downstream IL-6 signaling pathways with cytokine-driven hyperimmune reaction[59,69]. Compared to vehicle-treated animals, immunohistochemical analysis 4 days after the intervention, but not 8 weeks later, displayed a significant increase of CD68+ microglia (P < 0.01) and decrease of GFAP+ expression of astrogliosis in the lesion (P < 0.05), as well a reduced apoptosis with a significant decrease in cleaved caspase-3+ cells, compared to vehicle-treated SCI-rats. Eight weeks after these interventions, though, histological studies of the lesioned tissue in the Neuro-Cells and vehicle-treated SCI-rats did not establish any significant difference any more in the expression of microglia, astrocytes, and apoptosis. Compared to the baseline in vehicle-treated animals (set to 100%), proteomics in the Neuro-Cells-treated rats at that time still showed significant changes in the downregulation of pro-inflammatory proteins and the upregulation of the proteins involved in axonal and cellular regeneration [Figure 4]. An interesting finding was also the significant lower expression of Iba-1 in the spinal lesion of the Neuro-Cells-treated SCI-rats, compared to these animals treated with vehicle and/or intraperitoneal methylprednisolone, 10 weeks after these interventions [Figure 4].
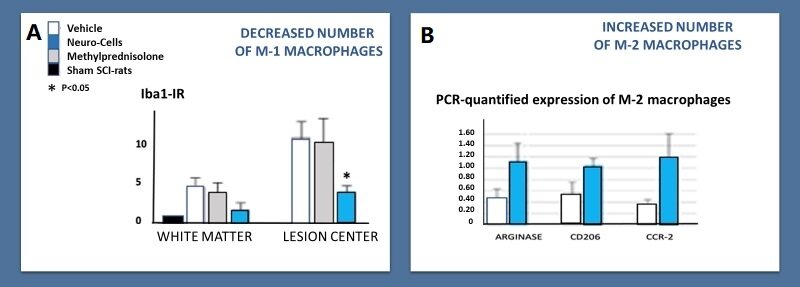
Figure 4. Changes in the M1/M2 paradigm 8-10 weeks after an intervention with bone marrow-derived stem cells (Neuro-Cells), methylprednisolone and vehicle in the acute phase of a spinal cord injury in rats. (A) The Western blot Iba1-IR quantified polarization from M1 to M2 microglia in the spinal cord white matter within the area close to and into the lesion center. Ten weeks after the intrathecal intervention with Neuro-cells, the Iba1 expression was significantly lower (P < 0.05) compared to the treatment with intrathecal vehicle and/or intra-peritoneal methylprednisolone 150 mg/kg (data normalized to Iba1-IR in the spinal white matter of intact healthy (sham) rats; statistical difference not indicated). Bars show means and SEM, n = 5-6 rats/group (Romero-Ramírez et al.[61], 2020, with permission of the authors). (B) Display of the increased levels of the typical M2-synthesized arginase-1 (inhibiting NO production), the M2 cell surface marker CD206 and the chemokine receptor CCR-2, polarizing macrophages toward an M2 phenotype, PCR-quantified polarization from M-1 to M-2 microglia (adapted from Wolters et al.[65] and de Munter et al.[71] with permission of the authors), 8 weeks after the acute intrathecal intervention of Neuro-cells in acute balloon compression-induced spinal cord injured rats, compared to SCI-rats, treated at the same time with only the vehicle. Due to the low numbers of experimental animals, significances were not reached.
Conforming to previous studies with stem cell implantations[70-72], intrathecal Neuro-Cells implants, but not interventions with riluzole and/or celecoxib in 10-week old asymptomatic FUS(1-358) and SOD1(G93A) mutant ALS-like mice were found to significantly delay motor dysfunction, as well as muscle atrophy and the loss of spinal lumbar motor neuron as seen in transgenic mice[61]. Interventions with Neuro-Cells in 12-week old asymptomatic FUS(1-358) frontotemporal lobe degeneration-like mice significantly delayed signs of depression and anxiety, cognitive deficits, and abnormal social behavior compared to FUS‐tg placebo-treated animals. Neuro-Cells did normalize prefrontal and hippocampal protein expression of IL‐1β, and of hippocampal Iba‐1 and GSK‐3β. In these transgenic mice, interventions with riluzole and celecoxib did bring the same beneficial effects, though way less pronounced[73].
SUMMARY AND CONCLUSION
Both in acute traumatic or hypoxic neurodegenerative lesions, as in chronic protein misfolding-induced neurodegenerative disorders, emerging evidence indicates that primary necrosis as induced by the underlying event initiates a secondary inflammatory process by a M1/M2 misbalance. This secondary process is responsible for a significant increase in the ultimate neurological deficit. These neurodegenerative diseases share a final common pathway, that is a M1/M2 misbalance-induced autoreactive response that targets against components of the nervous tissue.
Here, an up‐regulated protein expression of inflammatory markers, GSK‐3, regulating several signaling pathways including pro-inflammatory cytokine and interleukin production in the innate immune response, and Iba-1, a marker of microglia activation, reflects this degenerative process. These conditions also share an unmet need for disease-modifying interventions.
In this article, we presented data of preclinical studies after the effects of intrathecal implants of a human bone marrow preparation with truly naïve, negatively selected stem cells (Neuro-Cells) in rat models of spinal cord injuries, and in SOD-1 and FUS-transgenic amyotrophic lateral sclerosis-like and frontotemporal lobe degeneration-like mice. Neuro-Cells implants induced disease-modifying effects with changes in markers for M1/M2 macrophages/microglia, where (N)SAIDs failed. Indeed, both in immune-compromised and otherwise healthy experimental SCI-lesioned rats and in ALS- and frontotemporal lobe degeneration-like transgenic mice, intrathecal Neuro-Cells implants prevented for outrageous secondary inflammatory and apoptotic effects as evidenced by elevated GSK-3β and Iba-1 protein levels in the CNS. Therefore, it seems justified to further encourage clinical trials, applying bone marrow-derived naïve stem cells in patients suffering debilitating neurodegenerative diseases.
DECLARATIONS
AcknowledgementThe authors want to express their appreciation for the continuing interest and critical comments of Dr. Jorg Mey, Hospital Nacional de Parapléjicos, Toledo, Spain.
Authors’ contributionsParticipate in a close-knit international research team that performed the preclinical research discussed in this review article dealing with the effects of mesenchymal stem cells in neurodegenerative disorders: Wolters EC, Strekalova T, de Munter JPJM, Kramer BW
Availability of data and materialsNot applicable.
Financial support and sponsorshipNeuroplast BV, enabled by an innovation loan from the Ministry of Economic Affairs in The Netherlands, provided the bone marrow-derived human Neuro-Cells preparations, required in the various studies described in this paper. Beyond this sponsorship there was no additional funding involved.
Conflicts of interestIn addition to a staff position at the University of Maastricht, The Netherlands (Department of Neuroscience and Mental Health), Johannes de Munter is CEO of Neuroplast BV (the manufacturer of Neuro-Cells). Otherwise, there are no conflicts of interest whatsoever.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Hay KA, Hanafi LA, Li D, et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood 2017;130:2295-306.
4. Zhang C, Wu Z, Li JW, Zhao H, Wang GQ. Cytokine release syndrome in severe COVID-19: interleukin-6 receptor antagonist tocilizumab may be the key to reduce mortality. Int J Antimicrob Agents 2020;55:105954.
5. Chousterman BG, Swirski FK, Weber GF. Cytokine storm and sepsis disease pathogenesis. Semin Immunopathol 2017;39:517-28.
6. Shimabukuro-Vornhagen A, Godel P, Subklewe M, et al. Cytokine release syndrome. J Immunother Cancer 2018;6:56.
7. Fukui S, Iwamoto N, Takatani A, et al. M1 and M2 monocytes in rheumatoid arthritis: a contribution of imbalance of M1/M2 monocytes to osteoclastogenesis. Front Immunol 2017;8:1958.
8. Gust J, Hay KA, Hanafi LA, et al. Endothelial activation and blood-brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discov 2017;7:1404-19.
9. Subramaniam SR, Federoff HJ. Targeting microglial activation states as a therapeutic avenue in parkinson's disease. Front Aging Neurosci 2017;9:176.
10. Tang Y, Le W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol Neurobiol 2016;53:1181-94.
12. Poltavets AS, Vishnyakova PA, Elchaninov AV, Sukhikh GT, Fatkhudinov TK. Macrophage modification strategies for efficient cell therapy. Cells 2020;9:1535.
13. Villegas-Llerena C, Phillips A, Garcia-Reitboeck P, Hardy J, Pocock JM. Microglial genes regulating neuroinflammation in the progression of Alzheimer's disease. Curr Opin Neurobiol 2016;36:74-81.
14. Le W, Wu J, Tang Y. Protective microglia and their regulation in Parkinson's disease. Front Mol Neurosci 2016;9:89.
15. Politis M, Lahiri N, Niccolini F, et al. Increased central microglial activation associated with peripheral cytokine levels in premanifest Huntington's disease gene carriers. Neurobiol Dis 2015;83:115-21.
16. Hooten KG, Beers DR, Zhao W, Appel SH. Protective and toxic neuroinflammation in amyotrophic lateral sclerosis. Neurotherapeutics 2015;12:364-75.
17. Gómez-Nicola D, Schetters ST, Perry VH. Differential role of CCR2 in the dynamics of microglia and perivascular macrophages during prion disease. Glia 2014;62:1041-52.
18. Strachan-Whaley M, Rivest S, Yong VW. Interactions between microglia and T cells in multiple sclerosis pathobiology. J Interferon Cytokine Res 2014;34:615-22.
19. Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol 2015;15:545-58.
20. Schwartz MD, Emerson SG, Punt J, Goff WD. Decreased naïve T-cell production leading to cytokine storm as cause of increased COVID-19 severity with comorbidities. Aging Dis 2020;11:742-5.
21. Deczkowska A, Keren-Shaul H, Weiner A, Colonna M, Schwartz M, Amit I. Disease-associated microglia: a universal immune sensor of neurodegeneration. Cell 2018;173:1073-81.
22. Hammond TR, Dufort C, Dissing-Olesen L, et al. Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity 2019;50:253-71.e6.
23. Rustenhoven J, Jansson D, Smyth LC, Dragunow M. Brain pericytes as mediators of neuroinflammation. Trends Pharmacol Sci 2017;38:291-304.
24. Małkiewicz MA, Szarmach A, Sabisz A, Cubała WJ, Szurowska E, Winklewski PJ. Blood-brain barrier permeability and physical exercise. J Neuroinflammation 2019;16:15.
25. Brown LS, Foster CG, Courtney JM, King NE, Howells DW, Sutherland BA. Pericytes and neurovascular function in the healthy and diseased brain. Front Cell Neurosci 2019;13:282.
26. Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis 2010;37:13-25.
27. Ruan Q, Yang K, Wang W, Jiang L, Song J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med 2020;46:846-8.
28. Perrin P, Collongues N, Baloglu S, et al. Cytokine release syndrome-associated encephalopathy in patients with COVID-19. Eur J Neurol 2021;28:248-58.
29. Wu Y, Xu X, Chen Z, et al. Nervous system involvement after infection with COVID-19 and other coronaviruses. Brain Behav Immun 2020;87:18-22.
30. Mehta P, Mcauley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet 2020;395:1033-4.
31. Lucas C, Wong P, Klein J, et al. Yale IMPACT Team. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 2020;584:463-9.
32. Giamarellos-Bourboulis EJ, Netea MG, Rovina N, et al. Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell Host Microbe 2020;27:992-1000.e3.
34. Wang WY, Tan MS, Yu JT, Tan L. Role of pro-inflammatory cytokines released from microglia in Alzheimer's disease. Ann Transl Med 2015;3:136.
35. Su F, Bai F, Zhang Z. Inflammatory cytokines and Alzheimer's disease: a review from the perspective of genetic polymorphisms. Neurosci Bull 2016;32:469-80.
36. Park J, Han S, Mook-jung I. Peripheral inflammatory biomarkers in Alzheimer’s disease: a brief review. BMB Rep 2020;53:10-9.
37. Qin H, Buckley JA, Li X, et al. Inhibition of the JAK/STAT pathway protects against α-synuclein-induced neuroinflammation and dopaminergic neurodegeneration. J Neurosci 2016;36:5144-59.
38. Saitgareeva AR, Bulygin KV, Gareev IF, Beylerli OA, Akhmadeeva LR. The role of microglia in the development of neurodegeneration. Neurol Sci 2020;41:3609-15.
39. Wang Q, Liu Y, Zhou J. Neuroinflammation in Parkinson's disease and its potential as therapeutic target. Transl Neurodegener 2015;4:19.
40. Rocha NP, de Miranda AS, Teixeira AL. Insights into neuroinflammation in Parkinson's disease: from biomarkers to anti-inflammatory based therapies. Biomed Res Int 2015;2015:628192.
41. Qin XY, Zhang SP, Cao C, Loh YP, Cheng Y. Aberrations in peripheral inflammatory cytokine levels in Parkinson disease: a systematic review and meta-analysis. JAMA Neurol 2016;73:1316-24.
42. Hall S, Janelidze S, Surova Y, Widner H, Zetterberg H, Hansson O. Cerebrospinal fluid concentrations of inflammatory markers in Parkinson's disease and atypical parkinsonian disorders. Sci Rep 2018;8:13276.
43. Shen Z, Huang J, Wei H, et al. Validation of an in vivo electrochemical immunosensing platform for simultaneous detection of multiple cytokines in Parkinson's disease mice model. Bioelectrochemistry 2020;134:107532.
44. Rocha NP, Ribeiro FM, Furr-Stimming E, Teixeira AL. Neuroimmunology of Huntington's disease: revisiting evidence from human studies. Mediators Inflamm 2016;2016:8653132.
45. Chang KH, Wu YR, Chen YC, Chen CM. Plasma inflammatory biomarkers for Huntington's disease patients and mouse model. Brain Behav Immun 2015;44:121-7.
46. Yang HM, Yang S, Huang SS, Tang BS, Guo JF. Microglial activation in the pathogenesis of Huntington's disease. Front Aging Neurosci 2017;9:193.
47. Becher B, Spath S, Goverman J. Cytokine networks in neuroinflammation. Nat Rev Immunol 2017;17:49-59.
48. Paul A, Comabella M, Gandhi R. Biomarkers in multiple sclerosis. Cold Spring Harb Perspect Med 2019;9:a029058.
49. Poltavets AS, Vishnyakova PA, Elchaninov AV, Sukhikh GT, Fatkhudinov TK. Macrophage modification strategies for efficient cell therapy. Cells 2020;9:1535.
50. Wagner CA, Roqué PJ, Goverman JM. Pathogenic T cell cytokines in multiple sclerosis. J Exp Med 2020;217:e20190460.
51. Kwiecien JM, Dabrowski W, Dąbrowska-Bouta B, et al. Prolonged inflammation leads to ongoing damage after spinal cord injury. PLoS One 2020;15:e0226584.
52. Kozlowska U, Krawczenko A, Futoma K, et al. Similarities and differences between mesenchymal stem/progenitor cells derived from various human tissues. World J Stem Cells 2019;11:347-74.
53. Sivandzade F, Cucullo L. Regenerative stem cell therapy for neurodegenerative diseases: an overview. Int J Mol Sci 2021;22:2153.
54. Caplan AI. Mesenchymal stem cells: time to change the name! Stem Cells Transl Med 2017;6:1445-51.
55. Beers DR, Appel SH. Immune dysregulation in amyotrophic lateral sclerosis: mechanisms and emerging therapies. Lancet Neurol 2019;18:211-20.
56. Shahjin F, Chand S, Yelamanchili SV. Extracellular vesicles as drug delivery vehicles to the central nervous system. J Neuroimmune Pharmacol 2020;15:443-58.
57. Cofano F, Boido M, Monticelli M, et al. Mesenchymal stem cells for spinal cord injury: current options, limitations, and future of cell therapy. Int J Mol Sci 2019;20:2698.
58. de Munter J, Babaevskaya D, Wolters EC, et al. Molecular and behavioural abnormalities in the FUS-tg mice mimic frontotemporal lobar degeneration: Effects of old and new anti-inflammatory therapies. J Cell Mol Med 2020;24:10251-7.
59. Romero-Ramírez L, Wu S, de Munter J, Wolters EC, Kramer BW, Mey J. Treatment of rats with spinal cord injury using human bone marrow-derived stromal cells prepared by negative selection. J Biomed Sci 2020;27:35.
60. Gugliandolo A, Bramanti P, Mazzon E. Mesenchymal stem cells: a potential therapeutic approach for amyotrophic lateral sclerosis? Stem Cells Int 2019;2019:3675627.
61. de Munter JPJM, Shafarevich I, Liundup A, et al. Neuro-Cells therapy improves motor outcomes and suppresses inflammation during experimental syndrome of amyotrophic lateral sclerosis in mice. CNS Neurosci Ther 2020;26:504-17.
62. Andrzejewska A, Jablonska A, Seta M, et al. Labeling of human mesenchymal stem cells with different classes of vital stains: robustness and toxicity. Stem Cell Res Ther 2019;10:187.
63. Wolters ECh, de Hoo K, Kramer BW, de Munter JPJM. Anti-inflammatory effects of naïve stem cells dampen systemic/compartmental overreactive immune responses. J Immunol Sci 2021;5:37-43.
64. Chu F, Shi M, Zheng C, et al. The roles of macrophages and microglia in multiple sclerosis and experimental autoimmune encephalomyelitis. J Neuroimmunol ;2018,318:1-7.
65. Shen S, Sckisel G, Sahoo A, et al. Engineered IL-21 cytokine muteins fused to anti-PD-1 antibodies can improve CD8+ T cell function and anti-tumor immunity. Front Immunol 2020;11:832.
66. Jin MC, Medress ZA, Azad TD, Doulames VM, Veeravagu A. Stem cell therapies for acute spinal cord injury in humans: a review. Neurosurg Focus 2019;46:E10.
67. Oh KW, Noh MY, Kwon MS, et al. Repeated intrathecal mesenchymal stem cells for amyotrophic lateral sclerosis. Ann Neurol 2018;84:361-73.
68. Liau LL, Looi QH, Chia WC, Subramaniam T, Ng MH, Law JX. Treatment of spinal cord injury with mesenchymal stem cells. Cell Biosci 2020;10:112.
69. de Munter JP, Beugels J, Munter S, et al. Standardized human bone marrow-derived stem cells infusion improves survival and recovery in a rat model of spinal cord injury. J Neurol Sci 2019;402:16-29.
70. Uccelli A, Milanese M, Principato MC, et al. Intravenous mesenchymal stem cells improve survival and motor function in experimental amyotrophic lateral sclerosis. Mol Med 2012;18:794-804.
71. Boido M, Piras A, Valsecchi V, et al. Human mesenchymal stromal cell transplantation modulates neuroinflammatory milieu in a mouse model of amyotrophic lateral sclerosis. Cytotherapy 2014;16:1059-72.
72. Ciervo Y, Ning K, Jun X, Shaw PJ, Mead RJ. Advances, challenges and future directions for stem cell therapy in amyotrophic lateral sclerosis. Mol Neurodegener 2017;12:85.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Wolters EC, Strekalova T, Munter JPJM, Kramer BW. Naive BM-derived stem cells (Neuro-Cells) may modify acute and chronic neurodegenerative disorders by modulating macrophage behaviors. Ageing Neur Dis 2021;1:3. http://dx.doi.org/10.20517/and.2021.04
AMA Style
Wolters EC, Strekalova T, Munter JPJM, Kramer BW. Naive BM-derived stem cells (Neuro-Cells) may modify acute and chronic neurodegenerative disorders by modulating macrophage behaviors. Ageing and Neurodegenerative Diseases. 2021; 1(1): 3. http://dx.doi.org/10.20517/and.2021.04
Chicago/Turabian Style
Wolters, Erik Ch., Tatyana Strekalova, Johannes PJM de Munter, Boris W. Kramer. 2021. "Naive BM-derived stem cells (Neuro-Cells) may modify acute and chronic neurodegenerative disorders by modulating macrophage behaviors" Ageing and Neurodegenerative Diseases. 1, no.1: 3. http://dx.doi.org/10.20517/and.2021.04
ACS Style
Wolters, EC.; Strekalova T.; Munter JPJM.; Kramer BW. Naive BM-derived stem cells (Neuro-Cells) may modify acute and chronic neurodegenerative disorders by modulating macrophage behaviors. Ageing. Neur. Dis. 2021, 1, 3. http://dx.doi.org/10.20517/and.2021.04
About This Article
Copyright

Data & Comments
Data


 Cite This Article 25 clicks
Cite This Article 25 clicks

 Like This Article 32
likes
Like This Article 32
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.